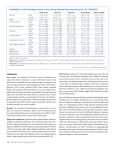
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Leukemia (2003) 17, 2358–2382 & 2003 Nature Publishing Group All rights reserved 0887-6924/03 $25.00 www.nature.com/leu REVIEW Treatment by design in leukemia, a meeting report, Philadelphia, Pennsylvania, December 2002 RA Larson1, GQ Daley2, CA Schiffer3, P Porcu4, C-H Pui5, J-P Marie6, LS Steelman7, FE Bertrand7 and JA McCubrey7,8 1 Section of Hematology/Oncology, University of Chicago Pritzker School of Medicine, Chicago, IL, USA; 2Whitehead Institute for Biomedical Research and Harvard Medical School, Cambridge, MA, USA; 3Wayne State University School of Medicine, Karmanos Cancer Institute, Detroit, MI, USA; 4Division of Hematology Oncology, Ohio State University College of Medicine and Public Health, Columbus, OH, USA; 5St Jude Children’s Research Hospital and the University of Tennessee Health Science Center, Memphis, TN, USA; 6Onco-Hematology Department, Hôtel-Dieu de Paris, Paris, AP-HP, France; 7Department of Microbiology and Immunology, The Brody School of Medicine at East Carolina University, Greenville, NC, USA; and 8Leo Jenkins Cancer Center, The Brody School of Medicine at East Carolina University, Greenville, NC, USA Novel approaches have been designed to treat leukemia based on our understanding of the genetic and biochemical lesions present in different malignancies. This meeting report summarizes some of the recent advances in leukemia treatment. Based on the discoveries of cellular oncogenes, chromosomal translocations, monoclonal antibodies, multidrug resistance pumps, signal transduction pathways, genomics/proteonomic approaches to clinical diagnosis and mutations in biochemical pathways, clinicians and basic scientists have been able to identify the particular genetic mutations and signal transduction pathways involved as well as design more appropriate treatments for the leukemia patient. This meeting report discusses these exciting new therapies and the results obtained from ongoing clinical trials. Furthermore, rational approaches to treat complications of tumor lysis syndrome by administration of the recombinant urate oxidase protein, also known as rasburicase, which corrects the biochemical defect present in humans, were discussed. Clearly, over the past 25 years, molecular biology and biotechnology has provided the hematologist/oncologist novel bullets in their arsenal that will allow treatment by design in leukemia. Leukemia (2003) 17, 2358–2382. doi:10.1038/sj.leu.2403156 Published online 9 October 2003 Keywords: leukemia therapy; signal transduction inhibitors; chromosomal translocations; multidrug resistance; monoclonal antibodies; rasburicase; elitekt; tumor lysis syndrome Introduction and overview of meeting: Richard A Larson, MD Several decades of prospective clinical trials have allowed the identification of important determinants of response for patients with leukemia that correlate with both treatment and survival. These have included age, the presence of comorbid illnesses, and the number of white blood cells (WBC) at time of diagnosis, and precise morphological diagnoses. Recently, more sophisticated techniques have been used to examine the biological characteristics of the leukemia itself including immunophenotyping, karyotyping and gene expression. This knowledge has led to risk-adapted treatment plans in which the use of particular cytotoxic drugs and supportive care measures have been designed for individual patients. Empiricism is gradually being replaced by individualized therapy. Targeted therapy did not start with STI571, now known as imatinib mesylate or Gleevect. The possibility of curing Correspondence: JA McCubrey, Department of Microbiology and Immunology, The Brody School of Medicine at East Carolina University, Brody Building 5N98C, Greenville, NC 27858, USA; Fax: þ 1 252 744 3104 Received 1 August 2003; accepted 26 August 2003; Published online 9 October 2003 disseminated cancer was ushered in approximately 50 years ago, when investigators recognized that inhibiting folate metabolism led to the death of childhood leukemia cells. Methotrexate, which inhibits dihydrofolate reductase and other enzymes, is still a cornerstone of treatment for acute lymphoblastic leukemia (ALL), although it has a minimal role in acute myeloid leukemia (AML). However, methotrexate is not selective. It inhibits the target enzymes in both normal and malignant cells. This symposium reviewed a number of pathways that are known to be important in the regulation of cell proliferation, apoptosis, drug resistance and metabolism in acute and chronic leukemia. Promising new anticancer drugs and the current status of clinical trials of investigational agents that have been designed to specifically inhibit or alter these pathways and interact with leukemia-specific targets were discussed at this meeting. This meeting report covers the role and requirement of the BCR-ABL oncoprotein in chronic myelogenous leukemia (CML), the transition from the chronic to the blast crisis stage and the permissive role for additional chromosomal translocations involving transcription factor genes and the roles of FLT-3 and RAS mutations in leukemia. The treatment of CML and other leukemias with Gleevect, and FLT-3 and RAS inhibitors is also discussed. Certain chromosomal translocations involve the expression of novel transcription factors that can exert dominant negative (DN) effects on gene expression by silencing transcription through chromatin remodeling by tethering histone decacetylases (HDAC). Treatment of certain leukemias with retinoic acids and HDAC inhibitors may prove efficacious. A frequent consequence observed in leukemia treatment with common anticancer drugs is drug resistance. Hematopoietic precursor cells may be intrinsically resistant to many drugs since they were ‘designed’ to survive repeated exposure to heterogeneous toxins and natural products. Drug resistance can arise by many different mechanisms; however, a common mechanism is increase in expression of a family of proteins called drug transporters (P-glycoprotein (P-gp), MRP-1, breast cancer resistance protein (BCRP) and others). These transmembrane proteins contain an ATP-binding cassette (ABC) and are associated with energy-dependent efflux of natural products. Many antileukemia drugs are substrates for multidrug resistance (MDR)-mediated efflux. P-gp, the protein product of the MDR-1 gene, is frequently overexpressed in stem cells. P-gp overexpression in AML is associated with a poorer prognosis. A means to circumvent MDR is the use of MDR inhibitors. Numerous MDR inhibitors have been developed and their Novel approaches to treat leukemia RA Larson et al 2359 effects are being evaluated in clinical trials (cyclosporine, PSC833, LY335979, VX710, XR9575, R101933, PK11195 and GG120918). The commonly used immunosuppressive drug cyclosporine is known to reverse the effects of P-gp. Chronic lymphocytic leukemia (CLL) is the most frequent leukemia observed in the Western Hemisphere. Historically, CLL has been treated with alkylating drugs. Now, however, fludarabine is the best single agent for initial therapy. Chimeric and humanized monoclonal antibodies (mAbs) represent novel therapeutic options for CLL. The humanized mAb targeting CD52 (CAMPATHs), a chimeric aCD20 Ab (Rituxans), an mAb targeting CD23 (IDEC 152) and a humanized mAb targeting an epitope of the HLA-DR b chain (Hu1D10, Remitogent) are in various stages of clinical trials in CLL patients. These modified antibodies may exert their cytotoxic effects by inducing apoptosis. ALL can be cured in approximately 80% of children and 30– 40% of adults. While up to 75% of ALL patients show identifiable genetic abnormalities associated with prognostic and therapeutic relevance, there remains considerable genetic heterogeneity, which can complicate effective treatment. Some high-risk ALL patients with certain chromosomal translocations (eg, BCR-ABL and MLL-AF4) can be effectively treated with allogeneic hematopoietic stem cell transplantation. The ability to classify ALL by genomic/proteomic approaches represents a significant advance in our understanding of the disease and will eventually provide the clinician with more precise therapeutic options for the patient. Tumor lysis syndrome (TLS) is a serious and potentially lifethreatening complication of cancer chemotherapy, especially in rapidly proliferating malignancies such as Burkitt’s lymphoma. TLS involves an increase in the levels of uric acid, potassium, phosphorous and other intracellular components due to the lysis of tumor cells. This increase in uric acid and other metabolites can lead to renal impairment primarily due to the insolubility of uric acid in the urine. Humans lack the ability to metabolize uric acid to allantoin, which is five times more soluble than uric acid, due to a mutation in the urate oxidase gene. For the past 30 years, urate oxidase has been purified from Aspergillus flavus and used to treat Burkitt’s lymphoma, ALL and AML patients in France and Italy. Rasburicase is a recombinant urate oxidase. Rasburicase is effective in preventing and treating hyperuricemia. Thus, this meeting covered various aspects of how basic scientific discoveries over the past 30 years have been applied clinically to improve significantly treatment of leukemia patients. Figure 1 Promising targets in leukemia: translating molecular mechanisms into rational therapies: George Q Daley, MD Dr Daley (Whitehead Institute and Harvard Medical School, Cambridge, MA, USA) discussed some of the scientific basis for rational drug design in leukemia. The dramatic success of Gleevec has galvanized the drug development community toward the identification of further such agents. This development has been facilitated by insights into the molecular mechanisms of cancer. There is an ongoing debate as to whether Gleevec will prove to be a rare example of great success or whether the principles responsible for Gleevec’s usefulness can be generalized to other tumor types. It is likely that our understanding of the rather limited set of pathways in cancer cells that lead to transformation can in many instances be targeted in the next decade or two. How many mutations are required to transform a normal cell into a malignant cell? This fundamental issue has been studied extensively in solid tumors by Vogelstein and colleagues at Johns Hopkins University (Baltimore, MD, USA). By studying the occurrence of various mutations in colon cancers, Vogelstein has derived a cancer model referred to by some as a ‘Vogelgram’, which assigns a sequence of stepwise mutations across the spectrum of colon cancer. It is estimated that at least four independent mutations must cooperate in the creation of a full-blown colonic malignancy.1,2 A diagram representing this concept is presented in Figure 1. Weinberg and colleagues at MIT modeled that work in primary cells,3 and recently it has become clear that at least three or four genetic mutations are required for the malignant transformation of primary cells. In initial rodent studies in the 1980s, it was shown that mutations at only two oncogenes myc plus ras were needed to generate a tumor cell.4 However, when those studies were attempted in human primary tissue, no tumors arose. It was not until the isolation of telomerase and the identification of its role in cellular immortalization did malignant transformation experiments in human cells start to work.5 Now it is clear that in primary human tissues, several collaborating oncogenes are required for malignant transformation. These genes fall into general categories, biological pathways or functional pathways that need to be deranged in all tumor cells in order for full tumorigenic phenotype to be realized. Immortalization and prevention of apoptosis need to be overcome. Genetic lesions that drive cell proliferation are Multiple mutations and time are required for the transformation of a malignant colon cancer cell. Leukemia Novel approaches to treat leukemia RA Larson et al 2360 necessary, and ras pathway mutations are common. Also, the problem of cellular senescence must be overcome and that is the function of the telomerase gene product. Standing in contrast to solid tumors, CML is an unusual disease that is classified as a myeloproliferative disorder. It is a weak malignancy, which some physicians consider a hyperplasia. The myeloproliferative phase of CML is associated with a consistent chromosomal abnormality recognized as a balanced translocation between distal elements of chromosomes 9 and 22.6 The human homologue of the Abelson murine leukemia virus oncogene, c-abl, is located on chromosome 9 and gets translocated and juxtaposed to a breakpoint cluster region on chromosome 22. This results in the Philadelphia chromosome (Ph), which encodes among other things the BCR-ABL oncoproteins.7 Over 10 years ago, Daley and colleagues at MIT inserted the BCR-ABL oncogene into a mouse retrovirus.8 Bone marrow cells of a mouse were infected with the recombinant retrovirus and a bone marrow transplant was performed. A CML-like myeloproliferative condition in the recipient mouse was observed. This led to the conclusion that BCR-ABL was indeed the causative genetic lesion driving CML. Leukemia It took a decade, however, to make this process of retroviral gene transfer and CML development more efficient, which confirmed that BCR-ABL, acting as a single agent, was in fact responsible for CML.9,10 It is known that every hematopoietic stem cell that gets infected by the BCR-ABL virus will contribute to the disease, and under experimental conditions in mice, a polyclonal disorder is observed. This suggests that at least in mice, the BCR/ABL oncoprotein alone can drive CML (Figure 2). This concept has now been generalized. There are many different chromosomal translocations that have been identified and characterized in the context of these hyperplasia-like myeloproliferative conditions. The translocation of 9 and 22, which juxtaposes BCR-ABL, is the best characterized and the most common, but it is now known that there are chromosomal translocations such as fusion of PDGF receptor and other genes such as TEL.11 TEL also fuses with ABL, and to other kinases like JAK.11 In general, the phenomenon seems to be that an activated kinase is responsible for driving the myeloproliferative phenotype. The global sites where these and other chromosomal translocations may act in the cell are presented in Figure 3. Genetic mutations that destroy the kinase activity of BCR-ABL abrogate all transforming functions of this oncoprotein. This Figure 2 Steps in transformation of human hematopoietic cells. Figure 3 Conversion from chronic to acute myelogenous leukemia requires at least two mutations. Novel approaches to treat leukemia RA Larson et al 2361 structure/function information provided the rationale for the development of the small molecule inhibitor Gleevec, which is a highly potent and specific inhibitor of the kinase activity developed by Novartis. If CML patients are treated with Gleevec immediately after diagnosis in the chronic phase, greater than 98% will achieve a complete normalization of the WBC counts.12–14 The vast majority of patients achieve a major cytogenetic response, that is, virtually complete suppression of evidence of the Ph in their cells. Clearly this is a dramatic result. The chronic phase of CML is unstable, and over time, these patients, left untreated, will progress to a malignancy typical of acute leukemia. This is called blast crisis. Blast crisis is the consequence of additional genetic abnormalities. This has been known at the cytogenetic level for many years, where patients who evolve blast crisis have an extra Ph, abnormalities of chromosome 17, trisomy 8, and a whole host of other superimposed genetic events. It thus appears that AML is at least a two-hit disease. This can be modeled in experimental murine systems, and supporting evidence has been gathered by cytogenetic analysis of clinical specimens. Patients have been described who present initially with the chronic phase of CML [t(9;22) bearing], and later evolve into blast crisis with leukemia cells bearing secondary genetic lesions such as the activation of homeobox genes or core-binding factor (CBF). This has also been seen in patients with myeloproliferative syndrome (MDS) characterized by the (5;12) translocation that evolved into AML with an additional (8;21) translocation. A concept promulgated by Dr Gary Gilliland (Harvard University, Boston, MA, USA) is that myeloproliferative conditions seem to evolve from activated kinases, whereas the progression to acute leukemia requires a block in differentiation that is typically seen in the context of translocations, which alter transcription factors.15 This concept has also been modeled in experimental systems. Gilliland et al have shown that BCR-ABL alone can induce CML. But if murine bone marrow cells are infected with a combination of viruses, one encoding BCR-ABL and the other carrying an altered transcription factor, AML is immediately observed in the transplanted mice.16 Gleevec is highly effective in chronic phase, perhaps because the chronic phase involves only a single hit. However, in blast crisis patients, Gleevec results in initial responses in virtually all patients, but a rapid relapse and resistance follows. Much of this resistance is due to mutations in the target BCR-ABL kinase12,17 indicating that the BCR-ABL remains an important target in this disease. Perhaps other drugs can be identified to target the mutant Gleevec-resistant forms of the BCR-ABL oncoprotein. It is also likely that there are other pathways activated in blast crisis. Therefore, the ultimate treatment will need target-directed agents against both BCR-ABL and the other activated pathways. Identification of these addition targets is crucial for complete CML therapy. Studies in AML have indicated the recurrent activation of or alteration of various transcription factors. The first and bestcharacterized translocation was the t(15;17) translocation in acute promyelocytic leukemia (APL), which juxtaposes the PML gene onto the retinoic acid receptor alpha (RARa) subunit gene.18–20 The most commonly targeted transcription factors in AML translocations include the RAR and the CBF, which has as its central DNA-binding component AML1 or RUNX-1, as well as various homeobox genes.21 The activity of RAR is a paradigm for the function of altered transcription factors in AML. The RAR acts as a dimer in concert with a partner, RXR. It sits on the transcriptional control region of a gene and acts to recruit a repressor complex.22–24 That repressor complex includes nuclear co-repressor proteins and principally the histone deacetylase complex (HDAC) that is responsible for modulating chromatin, leading to gene silencing.22–24 This repression can be overcome by the addition of retinoic acid (RA), which releases the repressor complex, and recruits an activator complex containing P300.25,26 RA acts as a potent differentiation agent in various hematopoietic cell types.27 The PML-RARa fusion protein encoded by the (15;17) translocation in APL binds the promoter and acts as DN inhibitors of transcription through stable transcriptional repression.28 The repressor complex binds to the promoter, blocking gene transcription and blocking expression of key genes involved in hematopoietic cell differentiation. This repressor complex is refractory to normal physiologic doses of RA. Pharmacologic doses of RA release the repressor complex, recruit the co-activator complex and restore the differentiation of blood cells.29 A diagram of the PML-RARa complex and the effects on gene transcription by RA and histone deacetylases is presented in Figure 4. This admittedly oversimplified model has nonetheless stimulated the idea that a target for this entire class of translocations is the transcriptional repressor complex. The most attractive target of this complex is the HDAC enzyme. Pharmacologic inhibition of HDAC has been shown to relieve the repressor complex, recruit the co-activator and restore hematopoietic differentiation.30,31 Recently, many HDAC inhibitors have been developed with micromolar potencies and introduced into clinical trials.32,33 In the laboratory, these have been shown to induce differentiation of a variety of hematopoietic cells, and there is encouraging clinical data in a growing variety of solid tumors as well as leukemias. Another emerging leukemia target is FLT3.34 FLT3 is a type 3/ class 3-receptor tyrosine kinase receptor that binds the growth factor FLT3 ligand, an important cytokine for the maintenance of hematopoietic stem cells. Naoe and colleagues have determined that about a quarter of AML patients has an internal tandem duplication of a juxtamembrane region of the receptor, which leads to its constitutive activation.35–38 Moreover, another cohort of AML patients harbor point mutations that involve the activation loop, a critical regulatory region around the enzymatic cleft.39 Therefore, almost a third of all patients with AML carry mutations in the FLT3 receptor in their leukemia cells. A number of FLT3 inhibitors are being evaluated in clinical trails that have previously been shown to be active against transformed cell lines and in mouse models.40,41 The clinical activity of these agents is among the most exciting new observations in target-directed treatment of AML. Another excellent pathway for targeting in leukemia is the Ras pathway. Ras is one of the canonical genetic lesions that is responsible for driving leukemia and solid tumor cell proliferation.42 The Ras pathway can be activated at many levels. Indeed, the FLT3 activating mutations themselves as well as essentially all mutations in tyrosine kinases are upstream of the Ras pathway, driving its activation and resulting in cell growth. A diagram of activation of the Ras pathway in leukemia is presented in Figure 5. For example, BCR-ABL can associate with adaptor proteins like Shc and Grb2 and activate Ras. Direct modulators of Ras activity, such as the Ras GAP proteins, can be mutated or activated in juvenile CML.43 There are many cases of myelodysplasia in AML, which have direct mutational activation Leukemia Novel approaches to treat leukemia RA Larson et al 2362 Figure 4 PML-RARa translocation in AML. (a) In a normal cell in the absence of RA, a repressor complex is present on certain genes that may modulate differentiation. (b) RA can induce a disassociation of transcription complexes on certain genes that may be involved in differentiation. (c) In cells containing PML-RARa chromosomal translocation, differentiation is inhibited as it acts as a DN transcription factor and displaces RXRa and secures HDAC. Figure 5 Activation of the RAS pathway in leukemia. of Ras. The Ras pathway can be activated in many ways and is thus an excellent therapeutic target. The enzymatic activity of Ras itself is not a particularly good target, but Ras is activated in an obligate way by a posttranslational isoprenylation.44 A fatty acid moiety is added to the C terminus of Ras by farnesyl transferase (FT). Ras is thereby targeted to cell membranes, and a number of companies have developed inhibitors of the FT that leave Ras in the cytosol as an inactive enzyme.45–47 BCR-ABL is known to activate the Ras pathway. Dr Daley’s laboratory has investigated whether farnesyl transferase inhibitors (FTIs) might serve as blockers of CML. In two different mouse models, his laboratory has shown that treatment of mice injected with BCR-ABL-transformed hematopoietic cells with Ras inhibitors led to enhanced survival.48 Moreover, in a BCRLeukemia ABL transgenic mouse model, the mice treated with FTIs had prolonged survival compared to vehicle-alone-treated mice.49 FTIs also have activity in hematopoietic colony-forming assays performed on cells taken from patient biopsies. BCRABL-positive colonies have a greater sensitivity to the drug than colonies from normal individuals.48 These results suggest there is therapeutic selectivity with FTI compounds, which could be useful in the treatment of CML. Together with Drs Mahon and Melo’s groups (Department of Haematology, Imperial College of Science, Technology and Medicine, Hammersmith Hospital, London, UK), Dr Daley has shown that FTIs are also effective against cells from patients who are resistant to Gleevec.50 Hematopoietic colony-forming assays indicate that treatment with one micromolar Gleevec leads to very low toxicity of BCR-ABL-positive cells from resistant Novel approaches to treat leukemia RA Larson et al 2363 patients. However, addition of an FTI at clinically achievable levels generates complete suppression, suggesting that resistance to Gleevec does not imply cross-resistance to FTI. It is therefore possible that FTI could be used successfully in combination with Gleevec in CML patients. Whether Ras is the true target of FTIs remains controversial. Ras is one of several hundred proteins in the cell that require post-translational modification by isoprenylation. There is now a large body of literature implicating various other targets within the cell that might be clinically relevant targets of FTIs.51–54 FTIs have been tested in early-stage clinical trials. Karp and colleagues at the University of Maryland Greenebaum Cancer Center (Baltimore, MD) have shown that the compound Zarnestras, which has an oral route of delivery, was effective in approximately 33% of the patients with highly refractory, drug-resistant AML.55 There were also some CML blast crisis patients in this study who had partial responses (PRs). Another FTI, lonafarnib, has also been tested in early phase clinical trials. Approximately 20–30% of patients had some clinical response with this compound. There has been some modest compassionate use of this compound together with Gleevec in some resistant patients. Combination clinical trials are being planned. A summary of where the several oncoproteins that have been discussed function in signal transduction pathway and where various inhibitors act is presented in Figure 6. In summary, understanding the total number of mutations in a cancer cell will provide insight into which target-directed agents are optimal for treating various malignancies. The CML-like syndromes appear to be the result of single genetic abnormalities and therefore target-directed agents are likely to have significant activity when used as monotherapy for these disorders. In contrast, the more acute malignancies, like acute leukemia, typified by CML in blast crisis, arise from at least two genetic lesions and perhaps more. Against acute leukemia, targetdirected agents are likely to be needed in combination. Dr Gilliland has developed a very nice heuristic model to promote this hypothesis. He classifies mutational lesions in AML as falling into two classes. Class I mutations, which typically drive cell proliferation, create a CML-like or myeloproliferative condition. These have been mimicked in mice by expression Figure 6 of BCR-ABL, TEL-PDGFR, and various activating mutations of FLT3 and the Ras pathway. Class I mutations exist in AML in concert with class II mutations, which typically lead to loss of function of hematopoietic transcription factors, perhaps through stable recruitment of HDAC or transcriptionally repressive complexes. Class II mutations act to block differentiation, and when seen alone in experimental models, induce an MDS-like condition. How many drugs will it take to treat malignancies that harbor multiple genetic mutations? If patients with the CML-like syndrome are treated early post diagnosis with targeted kinase inhibitors or perhaps the FTIs, significant single-agent activity can be observed. However, in the more genetically complex acute leukemias, combinations of target-directed agents will be needed for effective therapy. There is an enormous amount of enthusiasm because of the tremendous success of Gleevec. However, this enthusiasm should be tempered by the enormous challenges for targeted therapy that remain. It is daunting to consider the task of inhibiting multiple targets with highly specific agents. Each of these drugs will have to be put through the very complicated series of trials that are required before FDA approval. We are at the beginning of a very exciting era of target-directed leukemia therapy and once again, hematology seems to be paving the way for all of oncology in understanding how to translate molecular mechanisms into better therapies. Questions from audience Q: My question may be futile in this therapeutic new area, but how do you reconcile the one-hit basis of CML with Dr Fialkow’s data? George Daley, MD: The question is that there are classical data from Dr Fialkow and colleagues using clonality analysis that suggested in some patients that there might be a pre-existing clonal phase of hematopoiesis onto which the BCR-ABL translocation is superimposed.56 These data suggest that there may be pre-existing genetic lesions that contribute to the disease. I interpret the Fialkow data to suggest that a pre-existing tendency toward clonal expansion Site of action of oncogenes and signal transduction pathway inhibitors (black circles). Leukemia Novel approaches to treat leukemia RA Larson et al 2364 may indeed predispose individuals to the BCR-ABL translocation. CML is age-dependent and it tends to hit older rather than the younger individuals. So perhaps clonality represents a general precondition of the marrow, perhaps through decay of telomeres, which generates increased frequency of chromosomal translocations. It is by no means clear, however, that the Fialkow data imply a necessary prior genetic event. We know in model systems, at least in the mouse, that the single hit is functional. Emerging data from primary human cells generated by transferring BCR-ABL into primary human tissue indicate that a myeloproliferative phenotype can be seen in primary CD34 þ cells. Dr Fialkow’s data and the experimental data remain to be fully reconciled. Q: You covered most of the genes, which are involved in translocations. However, the ‘bad’ prognosis AML, for example, partial deletions of chromosomes 5 and 7, cannot be cured with chemotherapy. Even after bone marrow transplantation, the outcome is poor. What is known about critical genes that are involved in 5 and 7 chromosomal deletions? George Daley, MD: This is indeed a good point. Chromosomal deletion syndromes are frequently seen in myelodysplasias and secondary AMLs,57 and imply loss of tumor suppressor gene function. A general principle I also did not touch on is gene silencing – not just through transcriptional repression, but also through methylation.58 This leads to the therapeutic concept of demethylation agents, which may act to derepress or reactivate some of these classes of tumor suppressor genes. Q: Using very sensitive PCR, it is possible to demonstrate many of these translocations in totally normal individuals who never get leukemia. If one hit is enough, what is happening? George Daley, MD: It is actually quite alarming when one takes a sample of their own blood, and performs a sensitive PCR and finds BCR-ABL, which some in my lab have done. The reality is that the genetic lesion needs to occur in the right cell. The presumption is that as the hematopoietic stem cell undergoes dramatic proliferative expansion through all the various transit-amplifying populations, many translocations indeed occur. However, most of the target cells are unable to sustain the leukemia, because they are fated to ultimately differentiate, apoptose or die. So at least two bits of bad luck are needed for malignancy: the translocation has to occur in the first place and it has to occur in a cell that can sustain a long-term leukemia, like the hematopoietic stem cell. of eight or nine transmembrane protein pumps that contain ABCs.60–62 This is an energy-dependent process by which drugs that passively cross the cell membrane are very quickly pumped out. A number of these proteins have been characterized and may be clinically relevant in patients with AML. This meeting report will focus predominantly on MDR-1 or P-gp, because most of the in vitro and clinical work has been carried out with inhibitors of this particular pump. Many of the drugs used in the treatment of AML are substrates for MDR-1 and are rapidly extruded from drug-resistant cells.62,63 A diagram of the drug pumps and the effects of pump modulators is presented in Figure 7. Multidrug resistance in AML (Charles A Schiffer, MD) Drug resistance represents the major barrier to the successful treatment of leukemia, and AML in particular. Why is AML so resistant? First, AML is a disorder of hematopoietic precursors, which are designed to ‘live forever’ until failure occurs in other organ systems, such as the heart, kidneys or lungs. Excretory organs are in constant contact with multiple types of toxins, particularly from the organisms that we live with, and have developed mechanisms to survive such exposure. For example, most resistance mechanisms are amplified in gastrointestinal (GI) tract precursors. Similarly, bone marrow purging experiments indicate that hematopoietic stem cells can survive exposure in vitro to large doses of a variety of chemotherapeutic agents.59 Therefore, it is not particularly surprising that leukemias that derive from cells that resemble hematopoietic stem cells (eg, Ph-positive diseases, myelodysplasia and all the myeloproliferative diseases) are very resistant to chemotherapy. A major cause of resistance is a consequence of active efflux of drugs that have entered the cells, usually mediated by a group Leukemia Figure 7 Effects of drug pumps and modulators on chemotherapeutic drug resistance. Novel approaches to treat leukemia RA Larson et al 2365 Overexpression of P-gp can be detected in AML blasts at diagnosis, particularly in older patients, and may be further increased at relapse.63–67 In many studies, P-gp overexpression is associated with a poorer outcome, as is overexpression of some of the other efflux pumps, although the data on P-gp seem to be the strongest in this regard.64,66 Importantly, P-gpmediated resistance can be reversed in vitro by a variety of drugs, such as cyclosporine, PSC-833 and quinine.63,66 These in vitro observations led to a series of clinical trials evaluating modulators of drug resistance in patients with AML. A complicating issue is that the administration of some of the MDR1 inhibitors affects the metabolism and disposition of many chemotherapeutic drugs because of their effect on normal tissues such as biliary lining cells. In general, when modulators of these efflux pumps are given to patients, there is impaired excretion of the chemotherapeutic drug, increases in AUC and drug exposure and the potential for increased toxicity. In vitro assays of P-gp When in vitro assays on hematopoietic cells from AML patients are performed, they are usually performed on whole populations of leukemia cells. A problem, however, is that these cells are quite heterogeneous. For example, within a population of leukemia cells with similar morphology, there are dramatic differences in expression of different surface antigens. Furthermore, only a small fraction of the leukemia cells have the capacity to grow in long-term culture or in SCID mouse models. This subpopulation has been characterized by cell sorting as CD34 þ , HLA and DR negative. Animal experiments have shown that injection of this subpopulation of cells is capable of producing sustained tumors.68 Experiments with blasts from patients with AML have indicated that o1% of cells had the potential to produce long-term persistence either in culture or in animals, and might therefore represent leukemic ‘stem cells’ P-gp expression can be measured with antibodies directed against membrane P-gp epitopes by flow cytometric analysis or by measuring the function of the pump.69 Rhodamine 123 dye can be used to measure drug pump function as a surrogate for chemotherapeutic drugs. In ‘normals’, incubation with rhodamine results in increased fluorescence. In resistant cells however, there is decreased fluorescence because of rapid excretion of the dye. The effect of different modulators or pump inhibitors can also be evaluated in such assays. It can be difficult to detect drug resistance in a heterogeneous hematopoietic cell population. If only 0.5–1% of the cell population are actually responsible for persistence of leukemia after treatment, even very sensitive flow cytometric assays might be incapable of assessing the characteristics of this very small subpopulation of cells that may be most important in terms of drug resistance. To place this point in clinical perspective, even marginally effective drugs produce a multilog reduction in the number of AML cells that are circulating or in the bone marrow. Thus, it is the very small subpopulation of residual cells that are responsible for ultimate drug resistance and the patient’s death. Interpretation of in vitro assays of MDR can also be difficult. It has been known for years that there is variable and sometimes poor concordance among the immune, molecular and functional assays used to assess the effects of P-gp expression. Most laboratories now stress the importance of functional assays measuring the efflux/retention of the particular drug of interest. It is inferred in flow cytometry experiments that increased immunofluorescence representing increased chemotherapeutic drug retention results in increased cytotoxicity. However, a clinically relevant definition of ‘positive’ in any of these assays is not well defined and the magnitude of the in vitro fluorescence necessary to confer drug resistance in the patient is not known. Moreover, it is not clear which cells are killed – the clonogenic or the bystander cells. Lastly, it is not clear that the behavior of rhodamine, one of the substrates commonly used in drug efflux experiments, is similar to the effects of clinically important drugs such as the anthracyclines. It will take cumbersome clinical trials to answer these questions, which can take a long time and often can be difficult to analyze. Clinical trials with MDR modulators A number of clinical trials evaluating MDR modulators in AML have recently been completed. The CALGB completed a Phase I study, which was a prelude to a planned Phase III study, in older patients with previously untreated de novo AML.70 Patients received combination therapy with daunorubicin, etoposide and cytarabine with or without the MDR-1 modulator, PSC-833. The maximally tolerated dose without the modulator was 60 mg/m2 3 doses of daunorubicin, 100 mg/m2 3 doses of etoposide and 100 mg/m2 of cytarabine for 7 days. With the addition of PSC-833, however, the MTD were approximately 1/ 3 lower (40 mg/m2 of daunorubicin and 60 mg/m2 of etoposide) with mucositis as the dose-limiting toxicity. This was one of the largest Phase I trials that has ever been performed in acute leukemia, with results consistent with other studies demonstrating the necessity of dose reduction because of the effect of the MDR inhibitor on the pharmacokinetics (PK) of the chemotherapeutic agents. Trials using such modulators with reduced doses of chemotherapy are based on the hope that intracellular drug levels and resultant cytoxicity of the clonogenic cells will be increased to a much greater extent than can be achieved with higher doses alone. Indeed, earlier clinical trials suggest that higher doses of etoposide and/or daunorubicin are not sufficient to improve the outcome of treatment with AML. CALGB 9720 was an extension of the Phase I study in which older people with de novo AML were randomized to receive the two regimens described above.71 Unfortunately, this study was closed early because of excessive toxicity and mortality in the group receiving PSC-833. After about 120 patients were analyzed, there was a somewhat lower complete response rate in the experimental group, but a much higher early death rate associated with mucositis and infection. These results could not be explained by differences in the clinical characteristics in the two groups of patients. Overall, MDR-1 expression seemed to be a prognostic factor, in that the CR rate was higher in patients who were efflux-negative. The ECOG did a study evaluating PSC-833, which was also stopped early because of poor results. This study enrolled very high-risk patients including those in early relapse, with primary refractory disease, high-risk MDS or secondary leukemias and others who were post marrow transplantation. The patients were treated with a regimen of mitoxantrone, etoposide and ara-C with or without PSC-833.72,73 The doses of mitoxantrone and etoposide were reduced in the PSC-833 cohort. There was no difference in overall or disease-free survival, but because of a significant decrease in the CR rate in the PSC-833 arm, this study was stopped prematurely. In addition to the CALGB and ECOG studies, another large randomized trial conducted in Europe has been completed. While this study has not yet been published, it also failed to show an advantage of adding PSC-833 to induction therapy for older patients with AML. Leukemia Novel approaches to treat leukemia RA Larson et al 2366 Somewhat in contrast are the results of a study performed by SWOG using cyclosporine as an MDR modulator.73 PSC 833 is a nonimmunosuppressive, non-nephrotoxic analog of cyclosporine, which was much more active in vitro and hence was preferred by some groups in clinical trials. The SWOG study included high-risk patients with AML who received cytarabine and daunorubicin with or without cyclosporine. The same dose of daunorubicin was used in each arm, although a potentially important difference was that the daunorubicin was administered by continuous infusion rather than by the standard bolus used in the CALGB and ECOG studies. Responders received a second course of the same regimen. More than 200 patients were randomized, of whom more than half were in first relapse and almost 15–20% were primarily refractory. Among the patients who achieved remission, the relapse-free survival was significantly higher in the group of patients who received cyclosporine compared to the control group and what would have been predicted from historical experience. This translated into an overall survival advantage from the time of randomization for the cyclosporine recipients. What can be learned from these trials that can be applied in future studies and why have attempts to modulate P-gp in AML been disappointing? An obvious issue is that there are additional mechanisms of resistance not affected by PSC-833.73 An intriguing possibility is that cyclosporine might have an effect on these other resistance factors accounting for the positive results in the SWOG trial. There are also competing causes of treatment failure in AML, which present a design and statistical conundrum. At least a third of older AML patients will die of complications of treatment, which have nothing to do with whether MDR is successfully modulated. Although all patients should be included in the analysis of randomized trials, a high fraction of patients with early death dilute the ability to detect a biologic effect of a modulator of MDR. Furthermore, it is not known whether the modulator, even in vitro, enhances the killing efficiency of the critical clonogenic leukemia cells. Although it is possible to achieve levels in vivo that are effective in vitro, it is not known whether this occurs in every patient since there is considerable PK variability among individuals and higher levels may be needed in vivo to reverse drug resistance effectively. Another possibility is inadequate exposure to chemotherapy in the patients who are receiving reduced doses of chemotherapy with the expectation that the modulator will actually increase the drug exposure. If this PK effect does not occur reliably in every patient, it is possible that some individuals are actually being underdosed with chemotherapy with the potential for inferior outcome. AML may also not be the ideal tumor to evaluate the concept of MDR modulation. Only complete response is clinically relevant in AML, presenting a significant challenge for any new therapy. In contrast, in other diseases such as multiple myeloma or in certain solid tumors, partial remission is clinically very meaningful and may be more readily achievable. In addition, early death is not a major issue in such tumors. Lastly, clinical trials have focused on high-risk patients because there are lots of them and if a true benefit is observed, it could be very obvious and detected more rapidly than in patients with a better prognosis. However, these are also the hardest AML patients to treat, and small but clinically important differences could be missed particularly since such trials include a biologically heterogeneous population. There is also some disagreement about what the relevant end point should be. Obviously overall survival is critical, but some researchers have focused on the CR rate, or the fraction of patients who are cured rather than median survival. These issues have to be considered Leukemia when clinical trials are designed and affect sample size and study feasibility. Other MDR modulators There are a number of other drugs that affect MDR, which are in early clinical trials. These include Zosuquidar.HCl trihydrochloride (LY335979), which is reported not to have a PK effect on anthracycline disposition.74,75 Phase I trials in combination with standard dose chemotherapy have been completed and ECOG is contemplating a Phase III trial in AML patients utilizing this drug. Other compounds include VX710, which has the potential advantage of inhibiting MRP1 but which also had a significant effect on paclitaxel disposition and was associated with hyperbilirubinemia suggesting an effect on hepatic function.76,77 KXR9576 is an anthranilic acid derivative, which can be administered orally and intravenously.78,79 It has a prolonged effect after in vitro exposure to cells and it is presently in Phase I and II trials. In these studies, natural killer (NK) cells were isolated and characterized ex vivo after XR9576 infusion. NK cells normally overexpress MDR and in some patients, there was marked inhibition of rhodamine efflux after infusion of this MDR inhibitor, demonstrating a significant in vivo effect. R101933 is an MDR inhibitor that can be administered orally and intravenously. Side effects have included somnolence; no PK effect was demonstrated when used in combination with docetaxel.80 Lastly, GG120918 may have more pleiotropic effects in that it also modulates the BCRP.66 Future considerations What is next in drug resistance studies in AML? As noted, there are modulators with less PK effects, which are of considerable interest. It might be relevant to focus only on newly diagnosed younger patients, in whom an additional goal could be the prevention of the emergence of resistance. The CALGB has completed such a Phase I trial in younger patients with PSC-833 and has a randomized study in younger patients in progress. Pgp is also overexpressed in patients with acute lymphocytic leukemia, and there is enormous room for improvement in the treatment of adult ALL. Given the multiagent regimens used in ALL, however, ‘ideal’ trials with MDR modulators will be difficult to design. Finally, although a lot is known about drug resistance, little is known as to what promotes apparent drug sensitivity. For example, even in the poorest prognosis leukemias, a fraction of patients are cured with chemotherapy alone. The blasts from patients with acute progranulocytic leukemia, which are exquisitely sensitive to the effects of anthracyclines, have low expression of MDR1. Certain transcription factor mutations, TEL, AML1, AML1-ETO, can actually repress MDR expression by interacting with the promoter region of the MDR.81,82 These turn out to be highly curable leukemias. In other patients, differences in the cytokinetic state of the leukemic stem cell or immunemediated suppression of minimal residual disease may contribute to disease cure. It is hoped that greater understanding of these mechanisms of drug sensitivity and resistance will translate into improved outcomes in the future. Questions from audience Q: I just wanted to make a comment about biricodar. I should point out in the interest of fairness that I am working with the Novel approaches to treat leukemia RA Larson et al 2367 company that has tried to develop that drug. Your point about elevations in bilirubin is well taken; certainly if this occurs, it could affect the PK of anthracyclines. However, those elevations were very modest, and when one actually looks at very carefully done studies with PK of anthracycline alone and then in combination with biricodar, there are no substantial differences in the PK. These studies have also been extended to other types of drugs, such as mitoxantrone, and the results seem fairly consistent. So while I share your concerns, particularly in this very delicate population of patients, the results are encouraging. I have another point and I ask your thoughts about this. There have been a lot of questions raised about the cyclosporine results. I certainly view them as extremely encouraging. Is it conceivable that any of these results could occur by other effects of cyclosporine? I am curious about your views on this or do you think the data are done carefully enough to suggest that this occurs primarily by an MDR mechanism? Charles Schiffer, MD: With regard to the first comments, I thank you for those. I could not find literature or publications that showed that, but if indeed there is no effect on PK, particularly of the leukemia-relevant drugs, that is very important. I think it is easier to do a randomized study where you do not have to drop the dose of your chemotherapy in the experimental arm. Cyclosporine is a little more pleiotropic than we thought in terms of inhibition of other ABC pumps and that could be one explanation for the SWOG results, because it clearly had some benefit in that trial with patients who were not MDR-positive. Q: I have a question regarding secondary neoplasias, maybe as a consequence of MDR1 inhibition. You mentioned in the beginning of your talk the essential meaning of MDR1 in physiologic tissues. What about late consequences of MDR1 inhibition when anthracyclines or other toxic drugs are being administered? Charles Schiffer, MD: I must say, I have never thought about it. I have always assumed that these patients would be happy to have had that opportunity. In addition, the period of exposure is very brief. I guess, however, one could hypothesize that if you had an early intestinal lesion where MDR1 is an important component in the GI tract stem cell, that you might protect it in some way I am not aware of human or any animal data, but it is an interesting thought. Q: My question concerns the issue of stem cells and how important MDR is to the basic stem cells. When one examines the negative data – not the stuff by List, but the ECOG things you showed – is it lack of effect of the drug in PSC or is it the increased toxicity of the combination that results in the negative trial? I mean, is MDR so important in the basic stem cell that it is where the problem lies? Charles Schiffer, MD: I think it is both. With regard to the first question, there is increased toxicity and some increased mortality, and that becomes particularly evident if more fragile patients are examined. For example, in the CALGB study, in younger patients the toxicity was less of an issue. So I think it is a function of both. Recall that a positive trial in this disease is often obtained if the outcomes are improved by 10%. Oncologists would be ecstatic if that happened. However, with all the variables I have listed, one could see how even in large trials that have a couple of hundred patients, small differences could be obscured. This is why this has really been a very difficult field to study and to perform clean clinical trials. The evolving role of monoclonal antibodies in the treatment of CLL (Pierluigi Porcu, MD) CLL is the most common type of adult leukemia in the Western world.83 Most of the patients who develop CLL will need treatment at some point during the course of the disease, and many eventually will die from complications related to the disease or its treatment. CLL is not curable with conventional treatments. For a long time, CLL was considered a relatively simple and even unexciting form of leukemia, compared to the acute leukemias. This was more a reflection of the simplicity and limitations of the treatments available rather than lack of biological or clinical complexity. The complexity of CLL was initially recognized with the staging classifications of Rai and Binet. Greater therapeutic sophistication in CLL was first introduced with the independent discovery by Dr Grever (Ohio State University, Columbus, OH, USA) and Dr Keating (MD Anderson Cancer Center, Houston, TX, USA) that the purine analog fludarabine was able to induce significant responses in patients with refractory disease.84,85 Eventually, fludarabine was taken to front-line therapy with impressive results86 and is now being used alone or in combination with alkylating agents and monoclonal antibodies, producing a high number of complete responses. Drs Rai (Division of Hematology/Oncology, Long Island Jewish Medical Center, New Hyde Park, NY, USA) and Binet (Laboratory of Hematology, CHRU Bretonneau, Tours, France) recognized the existence of different prognostic groups of CLL patients.87–89 Even though the classifications are slightly different, both recognized that three major prognostic groups were with different survivals. More recently, advances in our knowledge of the molecular biology of CLL cells have led to the recognition of new biological prognostic markers, which can be integrated into the classifications of Drs Rai and Binet. Intriguing data about mutations in the variable region of the heavy immunoglobulin (Ig) chain (IgVH) in CLL cells have recently been reported.90,91 It is now recognized that there are two forms of CLL, one where the IgVH region contains somatic hypermutations (SHM), indicating that these cells are derived from germinal center (GC) B cells. These patients have a much better prognosis compared to patients with the second form of CLL, who do not have SHM in the IgVH region. A second type of biological risk stratification relies on the detection of specific chromosomal translocations in CLL cells.92–94 Three different subgroups of patients have been recognized who have remarkably different lengths of survival and treatment-free intervals (TFI), depending on which chromosomal aberration they carry. The presence of 17p deletion or 11q deletion are associated with clinically more aggressive disease and worse prognosis; patients with normal karyotype or trisomy 12 have an intermediate-risk prognosis; finally, patients with 13q deletion have a better outcome. A class of compounds that must be considered among targeted therapies is that of mAbs. They can be designed to target specific molecules important for the biology CLL cells, such as survival, proliferation and trafficking. A list of chimeric and humanized mAbs that are approved or in clinical development for the treatment of CLL is presented in Figure 8. Monoclonal antibodies have become a new paradigm in the treatment of hematologic malignancies.95,96 First, the toxicity is non-overlapping with that of chemotherapy and generally myelosuppression is not observed. They also have excellent biodistribution in key anatomical areas, such as the bone marrow, peripheral blood and spleen, that are critically involved in CLL and lymphoma. They have a novel, albeit incompletely Leukemia Novel approaches to treat leukemia RA Larson et al 2368 Figure 8 Use of engineered antibodies to treat CLL. understood, mechanism of action. Finally they have very broad spectrum of targets, such as surface antigens, cytokines, adhesion molecules, etc. One of the best-known monoclonal antibodies is rituximab (Rituxans), a chimeric antibody, which binds to the CD20 molecule expressed on B cell. It has significant single-agent activity in low-grade B-cell lymphoproliferative disorders and modest single-agent activity in diffuse large-cell lymphomas, with a very favorable toxicity profile. In CLL, early studies in previously treated patients using the standard weekly 4 schedule did not show a significant activity, but when previously untreated patients were examined it was recognized that standard-dose rituximab had reasonable activity.97 However, it is clear that, comparatively, standard-dose rituximab in CLL and small lymphocytic lymphoma (SLL) is substantially less active than in low-grade lymphomas. There may be different reasons for this phenomenon. One is the different PK of rituximab in CLL and SLL as opposed to lowgrade lymphomas. Other possibilities include a deficient or different mechanism of mAb-mediated killing compared to lowgrade lymphomas, resistance to complement-mediated killing and decreased CD20 density in CLL cells. Additionally, there may be a deficiency in immune-effector cells in CLL. These patients have significant underlying immunodeficiency related to their disease and they then are treated with alkylating agents, steroids and fludarabine, which add to the immunosuppression. Finally, defective signaling events leading to reduced sensitivity to apoptosis-inducing signals may also play a role. The problem of unfavorable PK has been addressed clinically in two different ways. O’Brien et al (Leukemia Department, The University of Texas MD Anderson Cancer Center, Houston, TX, USA) performed a dose-escalating trial with rituximab, which was taken up to 2.25 g/m2 as a weekly dose.98 This study showed that dose escalation could be accomplished safely, and that the response rates were increasing with higher doses of rituximab. A second approach was adopted by Byrd et al (Division of Hematology-Oncology, Ohio State University, Columbus, OH, USA), which used a three times a week administration of rituximab.99 This led to substantially higher Leukemia plasma levels of rituximab and also was associated with significant response rates in previously treated patients. These two studies illustrate how part of the resistance of CLL cells to rituximab might be overcome with higher plasma levels of antibody. Several questions remain, however, as to the specific mechanism of rituximab’s action in CLL as opposed to lowgrade lymphoma. One of the currently favored mechanisms of action of rituximab in B-cell malignancies is that it works through antibody-dependent cell cytotoxicity (ADCC). A significant amount of evidence suggests that ADCC is very important for the activity of rituximab. However, many of the antibodies that are currently used in the treatment of cancer are also known to induce intracellular signaling when tumor cells are examined in vitro, and the possibility that rituximab-induced signaling plays a role in its antitumor activity in vivo should be carefully examined. ADCC is mediated by a set of cell surface receptors that bind the Fc portion of the immunoglobulin G (FcgR). While many isoforms of FcgR exist, they can generally be distinguished as activating or inhibitory,100 depending on the effect that their engagement has on the ability of the effector cell to perform ADCC. Many innate immune effector cells carrying these receptors can efficiently mediate ADCC, remarkably among them NK cells, which only express the activating receptors.101 In contrast, other cells such as B cells and monocytes express some inhibitory receptor that can turn off ADCC. Jeffrey Ravetch’s group (Laboratory of Molecular Genetics and Immunology, Rockefeller University, New York, NY, USA), has very elegantly demonstrated the importance of FcgR in antibody-mediated tumor killing in vivo.102 They performed a study with mice infused with B16 melanoma cells. In wild-type mice given no treatment, massive pulmonary metastases were observed. If antibodies against the B16 melanoma cells were administered to the wild-type mice, a reduction of the incidence of pulmonary metastases was observed, although a substantial amount of disease was still present. However, when knockout mice for the inhibitory form, but not the activating form, of the FcgR were administered the antibody, there was a near- Novel approaches to treat leukemia RA Larson et al 2369 complete clearance of pulmonary metastases, indicating that in vivo the presence of inhibitory FcgR was decreasing the efficacy of the antimelanoma monoclonal antibodies. These experiments strongly suggest that in vivo, ADCC depends on the balance between inhibitory and activating FcgR and that alterations of this balance have profound effects on the outcome of therapy with mAbs. A way to increase the balance in favor of ADCC, with the goal of enhancing the efficacy of mAb-based therapy, is to increase the number of cells that are carrying the activating FcgR by giving a cytokine such as interleukin-2 (IL-2) that expends NK cells. Some of these approaches are being explored in lymphoma by many different research groups.103 On the other hand, Th2 cytokines such as IL-4 and IL-10, which increase the expression of inhibitory FcgR on these cells, are associated with a reduced ADCC effect. CLL, however, is a difficult disease target from the standpoint of ADCC for a number of reasons. First, ADCC usually requires a favorable effector-to-target ratio, which is very difficult to achieve in CLL because of the high number of circulating tumor cells. Second, patients with CLL tend to have a Th2 cytokine profile104–106 that downmodulates ADCC. Third, CLL cells express the inhibitory FcgR.107 Finally, experimental studies of ADCC in CLL are difficult to perform because there is no good preclinical model for human CLL at this time and it is hard to isolate sufficient effector cells from the peripheral blood to perform in vitro ADCC assays. Based on these considerations, one would predict that ADCC may not play a major role in the antitumor activity of rituximab in CLL. Induction of apoptosis may be an alternative possibility for the effect of rituximab in CLL.108 If CLL cells are incubated with rituximab in vitro, particularly when crosslinking antibodies are added, a substantial amount of apoptosis is observed. Evidence for a role of induction of apoptosis also comes from studies examining the signaling activity that follows rituximab binding to CLL cells in vivo. The proform of caspase-3 is converted into the active p16 form in cells taken from patients after a 1- to 3day exposure to rituximab.109 The level of antiapoptotic molecules such as XIAP and MCL-1 following exposure to rituximab is also reduced. Of note, caspase activation correlates with reduction in lymphocyte counts following rituximab therapy. A model for the mechanism of action of rituximab in CLL has therefore been proposed.109 Exposure to rituximab results in activation of the caspase pathway of apoptosis. Both caspase-3 and caspase-9 are activated. This results in activation of the downstream effectors of apoptosis. Additionally, two antiapoptotic molecules, MCL-1 and XIAP, are down-modulated in CLL cells following binding of the antibody, potentially making the cells more sensitive to the action of other drugs as well. What has been learned from the single-agent rituximab studies in CLL? First, rituximab is active as a single agent, particularly when the dose and the schedule of administration are targeted to reach a significant blood level of the antibody. Second, rituximab is safe, and excessive myelosuppression or infectious complications are not observed. Third, the apoptotic pathways activated in response to the binding of rituximab to CD20 are likely to have a major role in the antitumor activity of this antibody in CLL. How to improve on rituximab therapy? One obvious way is to combine the antibody with other active agents, such as fludarabine or alkylating agents. Another is to combine it with agents that down-modulate known survival signals for CLL cells coming from the microenvironment, among them tumor necrosis factor-a (TNF-a). To determine whether rituximab can be taken to the upfront stage in combination with fludarabine, CALGB conducted a clinical trial (CALGB 9712) where patients with newly diagnosed CLL were randomized to receive either fludarabine followed by rituximab or a concomitant regimen of fludarabine and rituximab followed by maintenance rituximab.110 A total of 104 patients were involved. A good number of them had intermediate risk and about half of them had high risk. The response analysis was intriguing, particularly in terms of the complete responses, indicating that there was a significant difference between the concurrent and the sequential arms, with 47% and 28% response rates respectively. These results suggest that rituximab was increasing the sensitivity of CLL cells to the cytotoxic effect of fludarabine. The other important observation in this trial was that this result was achieved at no cost of increased toxicity. In terms of the incidence of opportunistic infections, there were more opportunistic infections in the sequential as opposed to the concurrent arms. In terms of hematological and nonhematological toxicity, there was slightly more, but not significant, hematological toxicity in the concurrent arm. Some of the correlative studies performed by Dr Byrd et al in their initial trial of single-agent rituximab in CLL patients aimed at understanding the mechanism of the severe infusional reactions to rituximab, observed in CLL patients. Byrd et al observed that patients who were having severe infusional reactions had significantly higher levels of TNF-a as opposed to those who did not. TNF-a appears to be associated not only with toxicity, but it is also likely to be responsible for protecting CLL cells from apoptosis. TNF-a signaling through its receptor induces activation of NF-kB, which increases Bcl-2 transcription. This signal therefore not only increases survival, but also blocks the proapoptotic signaling induced by rituximab binding to CD20 in CLL cells. Therefore, TNF-a may become an attractive target from the therapeutic standpoint in addition to being a target in terms of reducing the infusional reactions. A clinical trial examining the combination of rituximab and etanercept, a dimeric fusion protein that binds TNF-a, blocking its interaction with cell surface receptors,111,112 is in progress at the Ohio State University. The hypothesis behind this trial is that by reducing the availability of TNF-a to CLL cells, there will be an increase in the efficacy of rituximab and potentially a reduction in the incidence of severe infusion reactions. Campath-1H is a humanized anti-CD52 antibody and has significant activity in T-cell prolymphocytic leukemia (T-PLL), CLL and cutaneous T-cell lymphomas (CTCL).113 The target antigen, CD52, is a GPI-linked glycoprotein whose function is not well known. CD52 is broadly expressed in B cells, T cells, monocytes, NK cells and dendritic cells.115 The mechanism of action of Campath-1H is not known. A clinical trial with Campath-1H was conducted in fludarabine-refractory patients.114–116 Campath-1H was administered intravenously by a stepped-up dosing with 3, 10 and 30 mg over a week and then followed by about 12 weeks of three times a week administration, with PCP and varicella zoster prophylaxis. In this pivotal trial, 92 patients were enrolled. The overall response rate was 33%. This was remarkable for fludarabine-refractory patients, who before the availability of Campath-1H had no viable option for therapy. The median survival was 16 months. Importantly, among the people who actually responded, there was a significant increase in median survival. Toxicity was substantial, both at the hematological and nonhematological level. A 50% incidence of grade 3/4 neutropenia and thrombocytopenia was observed. Infections, fever, rigors, rashes, nausea and vomiting, which were difficult Leukemia Novel approaches to treat leukemia RA Larson et al 2370 to manage, were also observed. Patients required antiemetics. Some deaths occurred from infection. An additional complication was frequent reactivation of cytomegalovirus (CMV). CALGB is now conducting two studies with Campath-1H. One consists of fludarabine induction, followed by randomization to intravenous Campath-1H vs a new subcutaneous (SQ) schedule of administration, which seems to be associated with a significantly lower incidence of infusional reactions. A second CLL trial in CALGB is CALGB10101, which is looking at the fludarabine–cyclophosphamide–rituximab (FCR) regimen to be followed by subcutaneous Campath-1H. In both of these trials, the principal goal is to achieve the highest possible level of complete remission in front-line patients with CLL. A third mAb in clinical trials for CLL is Hu1D10.117 Hu1D10 targets an epitope on the HLA-DR beta chain, which is expressed on many hematopoietic cells, including B cells, monocytes, T cells and dendritic cells. Altogether, about 50– 60% of B-cell lymphomas and approximately 75% of CLL patients express this epitope. Hu1D10 can mediate ADCC and complement-dependent cytotoxicity (CDC), and induce apoptosis in vitro.118,119 Similar to rituximab, when Hu1D10 is combined with crosslinking antibodies, the amount of cells that undergo apoptosis is significantly higher. Unlike rituximab, though, Hu1D10 does not activate caspase-3 or PARP cleavage. Hu1D10 reduces the mitochondrial membrane potential in CLL cells (JC Byrd, unpublished results), suggesting that the mechanism of induction of apoptosis in CLL cells with Hu1D10 might be different from that of rituximab. A Phase I study in lymphomas was recently completed. There were four drug levels with a standard weekly 4 schedule, stepping up from 0.15 to 5 mg/kg. In one cohort, there was an attempt to administer a more intensified treatment with daily incremental doses of Hu1D10 over a week. Some intriguing responses were observed in this Phase I trial in lymphomas, which led to a Phase II trial in low-grade lymphoma and an additional pilot Phase I trial in CLL and ALL. In the Phase II study in low-grade lymphoma, preliminary data do not appear to confirm the activity of this antibody. In CLL and in ALL, a Phase I study was initiated in collaboration between the Ohio State University and the University of Chicago. The schedule of administration was thrice weekly. This schedule was adopted because the antibody has a very short half-life. A stepped-up dosing was chosen, similar to the one used for Campath-1H, to decrease infusional toxicity. The study design was to gradually increase the dose of Hu1D10 to reach the target dose level for each particular cohort, which were 1.5, 3 and 5 mg/kg. In seven of 11 evaluable patients, there was a significant decrease in tumor volume at each dose level. Several patients had stabilization of their disease. There was a PR in a patient with a 17p deletion. This was very interesting, as these patients are known to be insensitive to rituximab. Moderate toxicity was observed with relatively unusual adverse events, such as delayed infusion toxicity with hypotension even several hours after the completion of the infusion, grade 3 hypophosphatemia, rash and urticaria. There were some cases of thrombocytopenia. Nausea was reported in these studies as well as headaches, which occurred in a significant number of patients. Based on the observation that Hu1D10-induced apoptosis in CLL cells follows different signaling pathways compared to rituximab (caspase-independent vs caspase-dependent, respectively) and the preliminary evidence of clinical activity in CLL, further development of this antibody in the treatment of CLL is being pursued. In conclusion, cytogenetics and IgVH mutational status are new powerful tools for risk stratifying CLL patients at diagnosis Leukemia and for treatment. This is a significant advance in terms of the planning of further clinical trials. The mAbs active in CLL may work differently in CLL as compared to non-Hodgkin’s lymphomas. Intriguing is the possibility that different mAbs may eliminate specific subsets of CLL clones, depending on which molecular abnormalities they carry. Therefore, combining different antibodies and biologic therapies should be the focus of the next generation of clinical trials. Questions from the audience Q: One of the hazards that seems to have emerged is the proximity of giving fludarabine followed by Campath-1H. A recent German trial has shown prolonged cytopenias accompanied by very profound fungal infections. Would you like to comment on that and how long a gap you really need to leave between the two? Pierluigi Porcu, MD: I would say that the approaches that are being taken right now are to wait several weeks, 4–6 weeks. I think that it also depends on the severity of myelosuppression following fludarabine, which may be significantly different from patient to patient. So, it is going to be difficult to have a homogenous approach that will be effective for all the patients. Q: I think the German trials are really quite worrying, because they did not leave even as much as 2 months, but there are several trials that are leaving 8 weeks. I am hesitant to say that is going to be enough, because they were getting 6 months of neutropenia after that combination. Pierluigi Porcu, MD: Yes, I agree that the issue of hematological toxicity is extremely important. Richard Larson, MD: I can comment on that, perhaps. In the CALGB study, there were only four cycles of fludarabine administered followed by a 2-month interval and then followed by 4 weeks of Campath-1H therapy. Fungal infections were not observed but reactivation of CMV occurred. Therefore, there is some concern about the combined immunotoxicity of that sequential regimen. Q: I think that the major challenge that we are facing now is when to treat early-stage A Binet CLL. I think that approximately 50% will never be treated and will live 20–30 years without any complications. So do you have any idea of the place of these antibodies in the treatment of CLL? Pierluigi Porcu, MD: I think that most of the combination approaches that are being explored now will have to be tested in Stage A Binet CLL patients who have high-risk disease defined by FISH or IgVH mutational status, as the toxicity of these combinations remains a big concern. This is only a minority of Binet A patients but one with no good expectations for a prolonged treatment-free interval or survival. I think that for patients who have good-risk early-stage CLL, deciding whether or not they should go on one of the clinical trials with combination of antibodies and fludarabine or antibodies, fludarabine, and alkylating agents remains a great challenge. A patient who might need therapy and is not willing to be treated on one of those trials, or has concerns about toxicity, could be treated with single-agent antibodies, since they are substantially more active in newly diagnosed patients. The remaining challenges in ALL (Ching-Hon Pui, MD) As cure rates in pediatric ALL reach 80%, emphases are placed on precise risk-directed therapy to avoid over- or undertreatment, to elucidate the mechanisms involved in drug Novel approaches to treat leukemia RA Larson et al resistance and to develop new therapeutic strategies.120 Genetic analyses have contributed greatly to our understanding of the pathogenesis and prognosis of ALL, and have begun to identify molecular targets for specific treatment.121 Heretofore, specific genetic abnormalities with prognostic relevance were limited to cases of B-cell precursor ALL. By use of DNA microarrays, Tom Look and colleagues (Harvard University, Boston, MA, USA) recently classified T-cell ALL into four major subtypes with prognostic significance: MLL-ENL and HOX11 clusters are associated with a favorable prognosis and TAL1 and LYL1 clusters with inferior outcome.122 Altogether, approximately 75% of childhood ALL cases now have identifiable specific genetic abnormalities with prognostic and therapeutic relevance.121 Although risk classification based on the primary genetic abnormalities of leukemic cells has great intuitive appeal, its predictive value is not especially high,123 probably because of secondary cooperative mutations, host pharmacogenetics and the over-riding impact of treatment on the prognostic information conveyed by these genetic abnormalities. For example, in an international collaborative study, Maurizio Aricò and associates showed that among Ph-positive ALL, an age of 1–9 years and a low presenting leukocyte count confer a relatively favorable prognosis.124 Ph-positive cases with a leukocyte count above 100 109/l have an extremely dismal outcome. More recently, the same group of investigators demonstrated the clinical heterogeneity among cases with 11q23 abnormalities (Table 1).125,126 Age was found to be the most important prognostic factor; infants younger than 1 year fared significantly worse than patients 1 year of age or older. Among patients with t(11;19) ALL and MLL-ENL fusion, those with a T-lineage immunophenotype, who were all over 1 year of age, had a superior outcome as compared to patients over 1 year of age with B-lineage ALL.126 Notably, there was heterogeneity even among infants with t(4;11) ALL; those with a poor early response to prednisone had an especially dismal prognosis. Gene expression profiling using DNA microarrays can not only accurately identify the major, important subtypes of ALL, but also provide insights into their underlying biology and the nature of cellular responses to antileukemic therapy.122,127–129 Dr James Downing and colleagues (St Jude Children’s Research Hospital, Memphis, TN, USA) studied 360 pediatric ALL patients. Distinct expression profiles identified six important leukemic subtypes, including T-cell ALL, E2A-PBX1, BCR-ABL, TEL-AML1, MLL rearrangement, and hyperdiploid 450 chromosomes.127 In addition, another ALL subgroup was identified based on its unique expression profile. Examination of the genes comprising the expression signatures provided important Table 1 The 5-year event-free survival estimates in ALL with 11q23 rearrangements, according to age and phenotype Type Category t(4;11) o1 year 41 year B-lineage, o1 year B-lineage, 41 year T-lineage o1 year 41 year o1 year 41 year o1 year 41 year t(11;19) t(9;11) del(11)(q23) Other 11q23 % EFS (s.e.) 19 42 27 46 88 38 46 40 73 22 65 (3) (5) (9) (14) (13) (15) (14) (22) (5) (8) (7) P-value o0.001 0.01 0.27 0.05 o0.001 2371 insights into the biology of these leukemia subgroups. For example, E2A-PBX1 leukemias were characterized by high expression of the c-MER receptor tyrosine kinase, a known transforming gene, suggesting that c-MER may be involved in the abnormal growth of these cells. Overexpression of FLT-3 receptor tyrosine kinase was detected in MLL-rearranged leukemias, a finding confirmatory of that recently reported by Stan Korsmeyer and colleagues.128 On the basis of this finding, a Phase I pediatric trial of inhibitor of FLT-3 tyrosine kinase will soon be initiated. To elucidate the genomics of cellular responses to cancer treatment, Dr William Evans and colleagues (St Jude Children’s Research Hospital, Memphis, TN, USA) analyzed the expression of over 9600 human genes in ALL cells before and after in vivo treatment with methotrexate and mercaptopurine given alone or in combination.130 Changes in expression of 124 genes accurately discriminated among the four treatments. Discriminating genes included those involved in apoptosis, mismatch repair, cell cycle control and stress response. Only 14% of genes that changed when these medications were given as single agents also changed when they were given together. Several gene clusters responded in a treatment-specific manner. For example, expression of the ataxia telangiectasia mutated (ATM) gene was higher after treatment with methotrexate alone but not after the other treatments. Consistent with G1 arrest by ATM, the percentage of ALL cells in S phase was lower after treatment with methotrexate alone but higher after the other three treatments. Ongoing studies by Dr Mary Relling and colleagues (St Jude Children’s Research Hospital, Memphis, TN, USA) are testing the hypothesis that gene expression profile can enhance our ability to identify patients at risk of developing specific complications. Preliminary data suggested that supervised hierarchical clustering and principal component analysis can identify selected genes expressed by ALL blasts that were associated with the development of secondary AML or brain tumors. However, these findings need to be confirmed by studying independent test sets. Inherited differences in the metabolism and disposition of drugs and polymorphisms in the genes that encode drugmetabolizing enzymes, transporters or targets can profoundly influence the efficacy and toxicity of drug therapy, thereby affecting the antileukemic response.131 Essentially all genes encoding drug-metabolizing enzymes responsible for key modifications of functional groups (phase I reactions) or for conjugation with endogenous substituents (phase II reactions) exhibit polymorphism. In general, phase I enzymes activate endogenous or exogenous substrates, and phase II enzymes deactivate or detoxify them. Thiopurine methyltransferase (TPMT) is a phase II enzyme that converts thiopurine prodrugs into methylated metabolites which are mostly inactive. This enzyme competes with other enzymes involved in the activation of thiopurine to their thioguanine nucleotides, which are incorporated into DNA and induce an antileukemic effect. TPMT activity is inherited as an autosomal codominant trait. Approximately 90% of the population are homozygous for the wild-type allele and have full enzyme activity; about 10% are heterozygous for the polymorphism and have intermediate levels of enzyme activity; and one of 300 persons carries two mutant TPMT alleles and does not express functional TPMT. Patients with TPMT deficiency accumulate higher levels of thioguanine nucleotides, and experience more hematopoietic toxicity when treated with standard doses of mercaptopurine, as compared to those with normal enzyme activity.132 Not surprisingly, patients who have deficient TPMT activity also tend to have better leukemic control than do those with normal Leukemia Novel approaches to treat leukemia RA Larson et al 2372 Leukemia activity.133 It should be noted that patients who have deficient TPMT activity are at an increased risk of epipodophyllotoxinrelated AML or irradiation-induced brain tumor, in the context of antimetabolite therapy.134,135 In fact, therapy-related AML has also been reported in these patients even when their treatment consisted primarily of antimetabolites.136 Thymidylate synthase, an essential enzyme in proliferating cells, is an important target of methotrexate. A homozygous triple-tandem-repeat polymorphism of the thymidylate synthase enhancer has been associated with increased enzyme expression and an inferior treatment outcome in childhood ALL.137 The homozygous polymorphism variant (substitution of T for C at position 677) of methylenetetrahydrofolate reductase, on the other hand, correlates with an increased risk of oral, GI or hepatic toxicity following low-dose methotrexate,138,139 and with greater in vitro sensitivity of leukemic blasts to methotrexate.140 Early response to therapy is one of the most important prognostic factors because it reflects both the leukemic-cell- and host-related characteristics. Measurement of MRD, by flow cytometric detection of aberrant immunophenotype or by PCR of clonal antigen-receptor gene rearrangements, affords a level of sensitivity and specificity not attainable by traditional morphologic assessment of treatment response.141,142 Dario Campana and colleagues (St Jude Children’s Research Hospital, Memphis, TN, USA) have shown that patients who attain a molecular or immunologic remission (defined as leukemic involvement of less than 0.01% of nucleated bone marrow cells) after 4–6 weeks of remission induction have a significantly better treatment outcome than do those who do not achieve this status.142,143 In fact, almost half of all patients have an MRD level p0.01% after only 2 weeks of remission induction and these patients have an exceptionally good prognosis, with less than 5% cumulative risk of relapse.144,145 By contrast, patients with an MRD level of X1% at the end of remission induction, a level that is difficult to detect by morphologic examination, fared as poorly as those who were considered induction failures by conventional criteria (ie, X5% marrow blast cells). Tandem application of flow cytometry and PCR testing allows us to successfully detect MRD in virtually 100% of cases of childhood ALL.146 We have therefore incorporated MRD detection into our current risk assessment system.120 Comparison of paired bone marrow and blood samples obtained concomitantly disclosed virtually identical levels from both sources in T-cell ALL but generally higher levels from bone marrow in B-lineage ALL.147,148 This finding suggested that T-cell ALL originates from extramedullary sites (eg, thymus) and subsequently invades bone marrow, and that T-cell cases can be monitored by examination of peripheral blood. While many advances have been made in the treatment of ALL, only CNS-directed therapy and hematopoietic stem cell transplantation will be briefly addressed here. Most contemporary clinical trials limit the use and dose of cranial irradiation because of concern over late sequelae. We recently reviewed the survival experience of our 10-year event-free survivors and observed an over 20% cumulative risk of secondary neoplasms at 30 years from diagnosis among those who had received CNS irradiation, resulting in a slight excess in mortality rate as compared to the general population.149 The irradiated patients also had a lower employment rate and among women, a lower marital rate. This finding should provide additional impetus to omit the use of cranial irradiation. Two studies have omitted cranial irradiation in all patients regardless of their risk classification, resulting in rates of isolated CNS relapse of 4.2 and 3% and of any CNS relapse of 8.3 and Table 2 ALL Risk factors for traumatic lumbar puncture at diagnosis of Risk factor Black Age o1 year Early eraa Less experience Platelet count o100 109/l a Odds ratio (95% CI) 1.5 2.3 1.4 1.4 1.5 (1.2–1.8) (1.7–3.0) (1.2–1.7) (1.1–1.8) (1.2–1.8) Lack of dedicated procedure area and general anesthesia. 6%, respectively.150,151 We believe that with more precise assessment of initial risk factors, and use of optimal intrathecal and systemic chemotherapy, CNS relapse hazard can be substantially reduced.152 In this regard, besides high-risk genetic features and large leukemic cell burden, the presence of leukemic cells in cerebrospinal fluid (even from iatrogenic introduction via traumatic lumbar puncture) is also associated with an increased risk of CNS relapse.153–155 We recently identified several factors associated with the risk of traumatic lumbar puncture, including experience of the clinician, platelet count and the use of sedation or anesthesia (Table 2).156 In our center, the lumbar puncture is now performed by the most experienced clinician and under deep sedation or general anesthesia. We transfuse all thrombocytopenic patients with platelets before lumbar puncture at diagnosis, and administer intrathecal chemotherapy immediately after collection of cerebrospinal fluid. Autologous transplantation has little if any therapeutic role in childhood ALL, and the indications for allogeneic stem cell transplantation from an HLA-matched related or unrelated donor during first remission of ALL are generally based on anecdotal evidence. A recent international collaborative study by Aricò and colleagues124 clearly demonstrated that allogeneic stem cell transplantation from an HLA-matched related donor is superior to intensive chemotherapy alone in prolonging initial complete remission in children with Ph-positive ALL, regardless of other risk features. However, transplantation did not improve outcome in patients with t(4;11).125,126 Studies are needed to determine definitively if transplantation benefits patients with severe hypodiploidy, near-diploidy or leukemia that has responded very poorly to initial remission-induction therapy. Several new agents are being tested. Arabinosylguanine is quite effective for T-cell ALL but it has narrow therapeutic index and is associated with neurotoxicity (eg, seizure and somnolence).157 Imatinib mesylate, which inhibits the BCR-ABL fusion protein and other tyrosine kinases, has induced a response rate of 70% with 20% complete (albeit transient) in patients with relapsed Ph-positive ALL,158 and will be tested in newly diagnosed cases. Other promising drugs include clofarabine159 and FLT-3 inhibitors.129 Further refinements in molecular classification, together with the identification of leukemic- and host-related genetic features that affect the efficacy and toxicity of antileukemic therapy, will afford unique opportunities to devise treatment plans for individual patients, and thus to further advance the cure rate. Questions from audience Q: I am curious about the data on the MRD. Do you think that will lead eventually to a change in how these patients are treated? There is the maintenance phase, which has the potential for toxicities. It is a heritage of the past where patients were Novel approaches to treat leukemia RA Larson et al 2373 treated for a very long period of time. Now we have some mechanisms to see whether or not their disease is there or not and do you think that this will be changed in the future. Ching-Hon Pui, MD: While MRD determination can identify patients at increased risk of relapse who may benefit from intensified therapy, I do not think it can be used to determine when we can stop therapy. ALL uniquely requires long-term continuation treatment. Although half of the patients have no measurable disease even by flow cytometry or PCR at the end of remission induction, all of them will relapse if continuation treatment is not given. The Tokyo Children’s Cancer Study Group had carried out an interesting study that featured only 1 year of therapy, and yielded an approximately 60% rate of long-term event-free survival. This finding indicates that 60% of patients may have therapy stopped at 1 year. However, this group of patients cannot be reliably distinguished from those who require prolonged therapy. Unfortunately, MRD determination cannot be used to guide cessation of therapy. For example, virtually all patients have negative MRD at the end of 212 years of continuation therapy but 10–20% of them will relapse. Therefore, MRD cannot be used to determine the length of continuation therapy. However, it may prove to be useful to determine the intensity of therapy. high lysozyme levels and dehydration. Dehydration is a frequent event in leukemia at diagnosis. All these abnormalities could be life threatening and obviously TLS has to be prevented or treated. A diagram of TLS and kidney dysfunction is presented in Figure 9. Different types of TLS are observed, for example, spontaneous TLS can occur before any chemotherapy in rapidly growing tumors and the best example is Burkitt’s lymphoma. Induced TLS is also observed in tumors highly sensitive to chemotherapy, like Burkitt’s, but also in acute leukemia. Tumor burden in acute leukemia can represent approximately 1 kg of tumor. After 1 week, 99% of the tumor is destroyed, which can create physiological disorders. Acute renal insufficiency is due to the renal tubular damage. Normally, phosphorus, uric acid and calcium are filtered by the glomerulus, and are partly reabsorbed in the proximal tubules. In the distal tubules the urine concentrates, and in the collecting tubules precipitation of uric acid could occur in case of hyperuricemia, especially due to the acidic pH of the urine. Also, precipitation of calcium phosphate can occur if the pH is basic. The situation is worsened when lysozymes also precipitate in tubules. This can occur in a patient with either a monocytic component or an M4 or M5 leukemia (according to FAB classification). This precipitation leads to acute tubulopathy, leading to anuria and acute renal failure. TLS in leukemias: frequency, diagnosis and management (Jean-Pierre Marie, MD) Frequency of TLS in adult acute leukemia TLS is a group of metabolic abnormalities associated with a rapid cell death in patients with cancer.160–187 All the contents of the cell will be released in the circulation when the cell dies. This means that released potassium could lead to hyperkalemia. Phosphorus release could lead to hyperphosphatemia and hypocalcemia due to phosphorus–calcium complexes, which are responsible for nephrocalcinosis. Hyperkalemia and hypocalcemia could be responsible for cardiac arrhythmia. The DNA present in the cell’s nucleus is degraded in the liver, which can give rise to hyperuricemia. Hyperuricemia and nephrocalcinosis are the major disorders associated with TLS; they could lead to acute renal insufficiency. This renal insufficiency could be worsened by other factors, including In the scientific literature it is very difficult to find an article on the frequency of TLS in adult acute leukemia. Dr Marie presented a retrospective analysis looking at TLS and its prevention or treatment in adults treated for acute leukemia during the last 3 years at the Hôtel Dieu Hospital in Paris. The files of 73 patients hospitalized at Hôtel Dieu hospital were reviewed. The mean age was 56. There were 63 AML and 10 ALL patients and the mean WBC count was 35 500. This is quite representative of the acute leukemia patients hospitalized at the Hôtel Dieu Hospital. The files of these patients were reviewed for TLS parameters. The levels of potassium, phosphorus, uric acid and creatinine were examined at the time of diagnosis before any chemotherapy to ascertain the possibility Figure 9 TLS and kidney dysfunction. This diagram provides an overview of the effects of TLS on kidney function and the effects of no treatment vs rasburicase treatment. Leukemia Novel approaches to treat leukemia RA Larson et al 2374 of spontaneous TLS. The patient files were reviewed also during the first week of intensive chemotherapy, to determine the frequency of chemotherapy-induced TLS. TLS at diagnosis Elevated life-threatening potassium levels were never observed. Significantly elevated phosphorus levels were observed in 15% of the cases. With regard to uric acid levels, 18% of the patients had a significant hyperuricemia. The mean values of creatinine for women and men according to EDGE were borderline. However, 26% of the patients presented with elevated creatinine levels at diagnosis. This could have resulted from dehydration. The study at Hôtel Dieu Hospital was unable to find any correlation between the WBC count and the potassium, phosphorus, creatinine or uric acid levels. But interestingly, there was a very good correlation between the creatinine and uric acid levels. A correlation between FAB subtype and creatinine levels was observed. Patients with a monocytic, monoblastic component (M4 and M5 of the FAB classification) had a higher creatinine level at diagnosis than the other myeloid leukemia patients. Lymphoblastic leukemia patients also had higher creatinine levels than the myeloid non-M4-M5 leukemia patients. The frequency of chemotherapy-induced TLS was examined. The variation of this parameter between day 0, before chemotherapy and during the first week of chemotherapy was examined. The percentage of variation of these parameters was examined. A mean variation of þ 11% was observed with potassium levels, but only four patients with more than a 50% increase in potassium levels were observed during the first week. A mean variation of þ 27% was observed with phosphorus, and 13% of patients had an increase in phosphorus levels of more than þ 50%. Phosphorus levels probably represent the best surrogate marker for TLS, because preventative treatments for decreasing phosphorus levels were not administered. On the other hand, a sharp decrease of uric acid levels with more than a 50% decrease was observed in the majority of the patients, and only 5% of patients experienced a 450% increase of serum uric acid level. With regard to the creatinine levels, the mean variation was not significantly different before and during chemotherapy, and two different patient populations were observed. A total of 42% of patients had a sharp (450%) decrease in creatinine levels. On the other end, 11% of the patients had a significant increase (450%) of their creatinine levels. Patients with uric acid and creatinine increases are the patients developing TLS most frequently. In summary, TLS in acute leukemia, according to this retrospective analysis, is present in about 20% of the patients at the time of diagnosis. A renal impairment was present in 26% of the patients, and uric acid and creatinine levels before the beginning of chemotherapy were highly correlated with TLS. Treatment of TLS Measures taken to contain TLS helped avoid major metabolic disorders during chemotherapy treatment. These measures consisted of vigorous hydration and alkalinization, control of the uric acid production as well as its degradation. Uric acid is a terminal product in human and primate catabolism of urine (Figure 10). Uric acid is a product of Leukemia oxidation of hypoxanthine and xanthine oxidase. Uric acid is not very soluble, and can crystallize in the urine. In other species, the terminal product is not uric acid but allantoin (Figure 10). Allantoin is the degradation product of uric acid by urate oxidase. Allantoin is five times more soluble than uric acid, avoiding a precipitation in the tubule. In humans, the urate oxidase protein is not functional because of a nonsense mutation. Prevention of uric acid overproduction relies on allopurinol Allopurinol is a very potent inhibitor of xanthine oxidase and it blocks the formation of uric acid156 (Figure 11). But this drug is unable to degrade the uric acid already formed. So it would be very useful to have a recombinant urate oxidase able to destroy uric acid and to transform it into allantoin avoiding renal uric acid crystallization. A nonrecombinant urate oxidase, Uricozymes, was extracted from Aspergillus flavus and has been available for over 30 years in France and Italy.188–194 It is widely used to avoid TLS in patients with Burkett’s lymphoma, ALL and AML. Severe allergic reactions and anaphylactic shock are rarely observed. But the limited production of Uricozyme and its subsequent purification for use in humans precluded its worldwide distribution. Recently, rasburicase, a recombinant urate oxidase, has become available in the USA and in Europe.160,189,195–201 In the USA, its use is restricted to pediatric patients with tumors. Compared to the nonrecombinant Uricozyme, this recombinant product has a greater purity and also a greater enzyme activity. The increased production of rasburicase has permitted worldwide distribution of its product. Rasburicase is much more active than allopurinol: 4 h after the first injection, there was a dramatic decrease of the serum uric acid levels after rasburicase compared to slow decrease after allopurinol. At 48 h, there was no difference in serum uric acid level between allopurinol and rasburicase. Concerning safety, in a European study of 107 patients, only 3% of adverse events related to the drug were observed. Two patients had rash and one patient had grade 3 hemolysis, but this patient was a G6PD-deficient patient. It was also observed that 7% of the patients generated antibodies (positive ELISA test), after the drug was administered. It is important to remember that urate oxidase is a foreign protein, not synthesized by humans. Three deaths were observed during the study, but none of them were related to rasburicase. The prevention of TLS in leukemic patients involves heavy hydration and antiuric acid treatment. A total of 77% of the patients in our series received three or more liters of hydration plus bicarbonate. Allopurinol was administered in 91% of the cases, often alone but sometimes with urate oxidase. In all, 29% of the patients received urate oxidase, sometimes alone but usually in combination with allopurinol. These measures to prevent hyperuricemia have to be initiated before chemotherapy in order to prevent increases in levels of creatinine during chemotherapy. When hyperuricemia was prevented before chemotherapy, only 10.5% of the patients showed elevated levels of creatinine during therapy. On the other hand, close to half of the patients displayed elevated levels of creatinine when the initiation of prevention and treatment of hyperuricemia were performed after the beginning of chemotherapy. Novel approaches to treat leukemia RA Larson et al 2375 Figure 10 Purine catabolism and prevention of TLS. This diagram depicts biochemical pathways responsible for degradation of purines following rapid cell lysis induced by chemotherapy and also illustrates the ability of either purified or recombinant urate oxidase to convert uric acid to allantoin. Figure 11 Allopurinol is an inhibitor of xanthine oxidase. This diagram depicts inhibition of xanthine oxidase by allopurinol to prevent conversion of hypoxanthine to xanthine to uric acid. Use of urate oxidase Urate oxidase was administered to two-thirds of the patients with increased creatinine levels at initial visit and to one-half of the patients with a high risk of TLS, such as FAB M4–M5 patients with lymphoblastic leukemia. The best time to begin urate oxidase treatment is before day 0 of chemotherapy, usually day 2 or day 3. Sometimes urate oxidase was administered later, but only as a salvage therapy when unexpected TLS developed. The levels of uric acid were examined 3 days after initiation of chemotherapy. A 90% decrease in uric acid levels was observed in patients treated with urate oxidase (7allopurinol), compared with a 36% decrease in patients treated with allopurinol alone. In summary, an early prevention of TLS before day 1 of chemotherapy reduces the risk of creatinine increases during chemotherapy. Allopurinol was administered to most patients, and urate oxidase was given preventively in an emergency setting to patients with monocytic components, in ALL patients with a large tumor burden, in patients with increased creatinine at presentation, and also in patients with high hyperleukocytosis needing an immediate chemotherapy within 1–2 days. According to those results, guidelines were proposed for the prevention and treatment of TLS in adult acute leukemia. In patients with a low risk of TLS, it is important to start hydration (1.5 l/m2 including bicarbonates) and allopurinol administration (600 mg/day) as soon as possible, usually 2 days before chemotherapy and until WBC decrease to normal. This Leukemia Novel approaches to treat leukemia RA Larson et al 2376 treatment is sufficient to control hyperuricemia and to prevent increase of creatinine levels in 75% of the cases. For patients who develop hyperuricemia despite this treatment, it is important to increase hydration and administer urate oxidase at 0.2 mg/kg/day until uric acid is definitively controlled. Usually, 2–3 days are enough to reduce uric acid levels to normal. In patients with high risk of TLS or even if TLS is already present or highly anticipated, it is important to begin as soon as possible before chemotherapy with vigorous alkaline hydration, 2–3 l/m2/day. Urate oxidase (0.2 mg/kg/day) is administered during the first few days of chemotherapy. In conclusion, TLS was found in roughly 20% of adult patients with acute leukemia. The main risk is acute tubulopathy, caused mainly by crystallization of uric acid. Urate oxidase catalyzes the conversion of uric acid to allantoin, avoiding tubular crystallization. TLS can be prevented through hydration plus antiuric acid treatment, mainly allopurinol or in selected patients, urate oxidase. Severe TLS has to be treated/prevented with vigorous hydration, and urate oxidase. Summary In this meeting report, exciting new therapies for leukemia treatment were discussed. Through molecular biology and cytogenetic approaches, many of the genes responsible for transformation of hematopoietic cells have been identified. Due to the elucidation that aberrant expression of certain oncoproteins causes cancer, fundamental increases in our understanding of the mechanisms of leukemogenesis have been obtained, which has yielded many novel therapeutic approaches. Many of these oncogenes activated in human cancer are created by the fusion of genes by chromosomal translocation. These translocations result in two broad classes of oncoproteins, kinases (eg, BCR-ABL, TEL-JAK) or transcription factors (TEL-AML-1, PMLRARa) with modified activities. Some oncogenes are activated by point mutations (RAS) or small amplifications of certain regions of the gene (FLT-3). Some chromosomal translocations (BCR-ABL, TEL/PDGFbR) may occur singly in myeloproliferative disorders; however, complimenting translocations must occur for the transition to acute leukemia (NUP98/HoxA9, AML1/ EVI1, AML1/ETO). More hope has been brought into the treatment of leukemia by the characterization of small molecular weight cell membrane permeable signal transduction pathway inhibitors. Gleevec is an inhibitor of BCR-ABL that can have astounding results on the chronic phase of CML with 495% normalization of blood counts. Other inhibitors have been developed that target the FLT-3 and Ras pathway (FLT-3, Raf and MEK inhibitors). These inhibitors may be especially useful in the treatment of CML patients whose leukemia cells have become resistant to Gleevec. Another class of inhibitors is represented by RA, which acts as a differentiating agent and can be effective in treating APL patients with PML-RAR chromosomal translocation. Furthermore, as more is known about how normal and chimeric transcription factors work, it has been clear that compounds that inhibit histone deacetylation (HDAC inhibitors) may prove effective in certain leukemia therapies. These compounds may prevent the block in differentiation induced by the chimeric transcription factor oncoproteins. Drug resistance remains a nagging problem in cancer chemotherapy. Eukaryotes have evolved pumps to remove toxins they encounter in their environments. Hematopoietic precursor cells may be particularly resistant to many chemotherLeukemia apeutic drugs, as they are ‘designed’ to survive repeated exposure to heterogeneous toxins. The overexpression of one of these pumps, P-gp, is detected in AML blasts, particularly in older patients, and may be increased at relapse. P-gp overexpression is associated with a poorer outcome. Modulators of the drug pumps have been discovered. These modulators (eg, cyclosporine, PSC-833) can reverse the effects of the drugresistant pumps. Thus, it is may be possible to treat patients with lower concentrations of the chemotherapeutic drug and the modulators. The goal of these studies with drug pump modulators is to raise the intracellular chemotherapeutic drug levels in the leukemic cells to a greater extent than can be achieved by administration of the chemotherapeutic drug by itself. A problem remains that these ‘drug pumps’ also serve important physiological roles in their normal tissues. Thus there is often toxicity associated with interference with the normal functions of the drug pumps. CLL is the most common type of leukemia observed in the Western Hemisphere with over 10,000 cases/year and is not curable with currently available therapy. Most patients are initially asymptomatic but eventually require therapy. Until the advent of monoclonal antibodies, the therapeutic options included treatment with chlorambucil, prednisone and purine analogs. Fludarabine is the best single drug for initial treatment and results in improved response rates and progression-free survival. However, there exists significant opportunities to improve fludarabine therapy. Humanized antibodies have been generated to treat CLL. The humanized antibodies have led to improved efficacy and diminished toxicity and represent new hope in the treatment of CLL. These humanized antibodies are directed at different proteins including CD22, CD23, CD52, HLA-DR and they exert their effects by different mechanisms including ADCC, CDC and apoptosis. Some of these antibodies will induce caspase 3-dependent (Rituxan) and caspase 3independent (Hu1D10) apoptosis. Hu1D10 induces apoptosis in CLL cells and it does not induce ADCC or CDC, and the apoptosis induced by Hu1D10 is caspase independent but leads to the production of AIF release, which may involve changes in the mitochondrial membrane potential. The apoptosis induced by Hu1D10 may occur by inhibition of Bcl-2 function leaving many of the other apoptotic machinery intact, as overexpression of Bcl-2 is protective and neutralizes the apoptotic effects of Hu1D10. Many other monoclonal antibodies have been developed to treat CLL and are currently being evaluated. Some of the antibodies target the CLL microenvironment and include aTNF, aVEGF, aIL4, aSDF and abFGF. Other antibodies are conjugated to toxins (aCD22–diptheria toxin, aCD19–genestein) or some are radiolabeled (aHLA-DR-I131, aCDC20Y90, aCD22, aCD52I131). There remain many challenges to improve the survival rate in ALL. The type of chromosomal translocation the patient has and the type of hematopoietic cell that the lesion is present in often determines how long a patient will have event-free survival. Leukemogenomics is now a real concept as gene expression profiling studies performed at St Jude Children’s Research Hospital have led to exciting methods that can classify patients into particular leukemia subtypes and can also predict whether those patients will have relapses over a given time period. This is also being coupled with determining the patient’s genotypes for genes involved in intermediary metabolism, drug metabolism, drug transport, drug receptor, host susceptibility and disease pathogenesis. Since many of these are enzymes involved in drug metabolism, these differences can affect the patient’s outcome after treatment with a particular drug. Thus leukemogenomics and pharmacogenetics will eventually provide the physician Novel approaches to treat leukemia RA Larson et al 2377 with the proper drug and concentration to treat individual leukemia patients. Complications of TLS can be prevented by either allopurinol or rasburicase treatment. This meeting also discussed the results of recent clinical trials designed to prevent TLS by either allopurinol or rasburicase treatment. Clearly, rasburicase is more effective and rapid in reducing uric acid levels resulting from TLS than allopurinol. As a result of the cloning of urate oxidase and the subsequent manufacturing of this recombinant protein, clinicians will be able to treat potential TLS patients with rasburicase. Proper identification of patients of high risk for TLS and their subsequent treatment with rasburicase will reduce renal problems associated with TLS. Adverse immune reactions resulting from administration of a foreign protein may develop if cancer patients are re-treated with rasburicase. However, this may not be a major problem if patients are cured of their cancer after a single treatment. Rasburicase is an intriguing example of how basic science can be applied to prevent a disease. A recombinant enzyme absent in humans is infused to catabolize uric acid that could otherwise accumulate to toxic levels in certain disease states. Cancer research has surpassed many important milestones in the past 30 years. These advances have stemmed from technological discoveries that have allowed the cloning of DNA sequences, the discovery of oncogenes and their roles in human cancer, the knowledge of how transcription factors, promoters and chromatin structure affect gene expression, the sequencing of the human genome, and the discovery and clinical applications of monoclonal antibodies. Many scientific advances have been achieved by brute-force screening of pharmaceutical libraries. This has led to numerous clinical trials, which have tested the effects of particular compounds or antibodies to suppress cell growth, which might under certain circumstance cure the leukemia patient. Undoubtedly and most often, these drug screening and clinical trials yielded negative results. But we must always be optimistic with the advances that the cancer field has made, and continue to test possible mechanism and therapies. Unquestionably, advances have been made that have improved the quality of care and the extended survival of leukemia patients. Acknowledgements GQ Daley was supported in part by the Grants NIH CA76418, CA86991, DK59279 and HL71265, and a sponsored research agreement from the Schering-Plough Research Institute. GQ Daley is a Birnbaum Scholar of the Leukemia and Lymphoma Society of America. P Porcu was supported in part by a grant (K23CA102155) from the NCI. C-H Pui was supported in part by the Grants CA 21765, CA 31566, CA 51001, CA 78824, CA 29139, CA 37379 and GM 61393 from the US National Institutes of Health; a Center of Excellence grant from the State of Tennessee, USA; and the American Lebanese Syrian Associated Charities. C-H Pui is the American Cancer Society FM Kirby Clinical Research Professor. JA McCubrey was supported in part by the Grants CA 51025 and CA098195. References 1 Cho KR, Vogelstein B. Genetic alterations in the adenoma – carcinoma sequence. Cancer 1992; 70 (6 Suppl): 1727–1731. 2 Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990; 61: 759–767. 3 Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100: 57–70. 4 Land H, Parada LF, Weinberg RA. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 1983; 304: 596–602. 5 Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature 1999; 400: 464–468. 6 Deininger MWN, Goldman JM, Melo JV. The molecular biology of chronic myeloid leukemia. Blood 2000; 96: 3343–3356. 7 Ben-Neriah Y, Daley GQ, Mes-Masson AM, Witte ON, Baltimore D. The chronic myelogenous leukemia-specific P210 protein is the product of the bcr/abl hybrid gene. Science 1986; 233: 212–214. 8 Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science 1990; 247: 824–830. 9 Pear WS, Miller JP, Xu L, Pui JC, Soffer B, Quackenbush RC et al. Efficient and rapid induction of a chronic myelogenous leukemialike myeloproliferative disease in mice receiving P210 bcr/abltransduced bone marrow. Blood 1998; 92: 3780–3792. 10 Li S, Ilaria Jr RL, Million RP, Daley GQ, Van Etten RA. The P190, P210, and P230 forms of the BCR/ABL oncogene induce a similar chronic myeloid leukemia-like syndrome in mice but have different lymphoid leukemogenic activity. J Exp Med 1999; 189: 1399–1412. 11 Look AT. Oncogenic transcription factors in the human acute leukemias. Science 1997; 278: 1059–1064. 12 Azam M, Latek RR, Daley GQ. Mechanisms of autoinhibition and STI-571/imatinib resistance revealed by mutagenesis of BCR-ABL. Cell 2003; 112: 831–843. 13 Nagar B, Hantschel O, Young MA, Scheffzek K, Veach D, Bornmann W et al. Structural basis for the autoinhibition of c-Abl tyrosine kinase. Cell 2003; 112: 859–871. 14 O’Brien SG, Guilhot F, Larson RA, Gathmann I, Baccarani M, Cervantes F et al. Imatinib compared with interferon and lowdose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 2003; 348: 999–1004. 15 Deguchi K, Gilliland DG. Cooperativity between mutations in tyrosine kinases and in hematopoietic transcription factors in AML. Leukemia 2002; 16: 740–744. 16 Dash AB, Williams IR, Kutok JL, Tomasson MH, Anastasiadou E, Lindahl K et al. A murine model of CML blast crisis induced by cooperation between BCR/ABL and NUP98/HOXA9. Proc Natl Acad Sci USA 2002; 99: 7622–7627. 17 Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2002; 2: 117–125. 18 Kakizuka A, Miller Jr WH, Umesono K, Warrell Jr RP, Frankel SR, Murty VV et al. Chromosomal translocation t(15;17) in human acute promyelocytic leukemia fuses RAR alpha with a novel putative transcription factor, PML. Cell 1991; 66: 663–674. 19 de The H, Lavau C, Marchio A, Chomienne C, Degos L, Dejean A. The PML-RAR alpha fusion mRNA generated by the t(15;17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell 1991; 66: 675–684. 20 Goddard AD, Borrow J, Freemont PS, Solomon E. Characterization of a zinc finger gene disrupted by the t(15;17) in acute promyelocytic leukemia. Science 1991; 254: 1371–1374. 21 Pandolfi PP. Oncogenes and tumor suppressors in the molecular pathogenesis of acute promyelocytic leukemia. Hum Mol Genet 2001; 10: 769–775. 22 Fenrick R, Hiebert SW. Role of histone deacetylases in acute leukemia. J Cell Biochem Suppl 1998; 30-31: 194–202. 23 Lin RJ, Nagy L, Inoue S, Shao W, Miller Jr WH, Evans RM. Role of the histone deacetylase complex in acute promyelocytic leukaemia. Nature 1998; 391: 811–814. 24 Grignani F, De Matteis S, Nervi C, Tomassoni L, Gelmetti V, Cioce M et al. Fusion proteins of the retinoic acid receptor-a recruit histone deacetylase in promyelocytic leukaemia. Nature 1998; 391: 815–818. 25 Chen A, Licht JD, Wu Y, Hellinger N, Scher W, Waxman S. Retinoic acid is required for and potentiates differentiation of Leukemia Novel approaches to treat leukemia RA Larson et al 2378 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 Leukemia acute promyelocytic leukemia cells by nonretinoid agents. Blood 1994; 84: 2122–2129. Lin RJ, Sternsdorf T, Tini M, Evans RM. Transcriptional regulation in acute promyelocytic leukemia. Oncogene 2001; 20: 7204– 7215. Collins SJ. The role of retinoids and retinoic acid receptors in normal hematopoiesis. Leukemia 2002; 16: 1896–1905. Lin RJ, Sternsdorf T, Tini M, Evans RM. Transcriptional regulation in acute promyelocytic leukemia. Oncogene 2001; 20: 7204– 7215. Doucas V, Brockes JP, Yaniv M, de The H, Dejean A. The PMLretinoic acid receptor a translocation converts the receptor from an inhibitor to a retinoic acid-dependent activator of transcription factor AP-1. Proc Natl Acad Sci USA 1993; 90: 9345–9349. Minucci S, Nervi C, Lo Coco F, Pelicci PG. Histone deacetylases: a common molecular target for differentiation treatment of acute myeloid leukemias? Oncogene 2001; 20: 3110–3115. Melnick A, Licht JD. Histone deacetylases as therapeutic targets in hematologic malignancies. Curr Opin Hematol 2002; 9: 322– 332. Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov 2002; 1: 287–299. Kelly WK, O’Connor OA, Marks PA. Histone deacetylase inhibitors: from target to clinical trials. Expert Opin Invest Drugs 2002; 11: 1695–1713. Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood 2002; 100: 1532–1542. Kiyoi H, Ohno R, Ueda R, Saito H, Naoe T. Mechanism of constitutive activation of FLT3 with internal tandem duplication in the juxtamembrane domain. Oncogene 2002; 21: 2555–2563. Hayakawa F, Towatari M, Kiyoi H, Tanimoto M, Kitamura T, Saito H et al. Tandem-duplicated Flt3 constitutively activates STAT5 and MAP kinase and introduces autonomous cell growth in IL-3-dependent cell lines. Oncogene 2000; 19: 624–631. Kiyoi H, Towatari M, Yokota S, Hamaguchi M, Ohno R, Saito H et al. Internal tandem duplication of the FLT3 gene is a novel modality of elongation mutation which causes constitutive activation of the product. Leukemia 1998; 12: 1333–1337. Yokota S, Kiyoi H, Nakao M, Iwai T, Misawa S, Okuda T et al. Internal tandem duplication of the FLT3 gene is preferentially seen in acute myeloid leukemia and myelodysplastic syndrome among various hematological malignancies. A study on a large series of patients and cell lines. Leukemia 1997; 11: 1605–1609. Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R, Kodera Y, Miyawaki S et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood 2001; 97: 2434–2439. Tse KF, Novelli E, Civin CI, Bohmer FD, Small D. Inhibition of FLT3-mediated transformation by use of a tyrosine kinase inhibitor. Leukemia 2001; 15: 1001–1010. Levis M, Tse K-F, Smith BD, Garrett E, Small D. A FLT3 tyrosine kinase inhibitor is selectively cytotoxic to acute myeloid leukemia blasts harboring FLT3 internal tandem duplication mutations. Blood 2001; 98: 885–887. Shannon K. The Ras signaling pathway and the molecular basis of myeloid leukemogenesis. Curr Opin Hematol 1995; 2: 305–308. Largaespada DA. Genetic heterogeneity in acute myeloid leukemia: maximizing information flow from MuLV mutagenesis studies. Leukemia 2000; 14: 1174–1184. Pérez-Sala D, Rebollo A. Novel aspects of Ras proteins biology: regulation and implications. Cell Death Differ 1999; 6: 722–728. Adjei AA. Blocking oncogenic Ras signaling for cancer therapy. J Natl Cancer Inst 2001; 93: 1062–1074. Le DT, Shannon KM. Ras processing as a therapeutic target in hematologic malignancies. Curr Opin Hematol 2002; 9: 308–315. Chang F, Steelman LS, Lee JT, Shelton JG, Navolanic PM, Blalock WL et al. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: potential targeting for therapeutic intervention. Leukemia 2003; 17: 1263–1293. Peters DG, Hoover RR, Gerlach MJ, Koh EY, Zhang H, Choe K et al. Activity of the farnesyl protein transferase inhibitor SCH66336 against BCR/ABL-induced murine leukemia and primary cells from patients with chronic myeloid leukemia. Blood 2001; 97: 1404–1412. 49 Reichert A, Heisterkamp N, Daley GQ, Groffen J. Treatment of Bcr/Abl-positive acute lymphoblastic leukemia in P190 transgenic mice with the farnesyl transferase inhibitor SCH66336. Blood 2001; 97: 1399–1403. 50 Hoover RR, Mahon FX, Melo JV, Daley GQ. Overcoming STI571 resistance with the farnesyl transferase inhibitor SCH66336. Blood 2002; 100: 1068–1071. 51 Lebowitz PF, Prendergast GC. Non-Ras targets of farnesyltransferase inhibitors: focus on Rho. Oncogene 1998; 17: 1439–1445. 52 Du W, Lebowitz PF, Prendergast GC. Cell growth inhibition by farnesyltransferase inhibitors is mediated by gain of geranylgeranylated RhoB. Mol Cell Biol 1999; 19: 1831–1840. 53 Law BK, Nørgaard P, Gnudi L, Kahn BB, Poulson HS, Moses HL. Inhibition of DNA synthesis by a farnesyltransferase inhibitor involves inhibition of the p70S6k pathway. J Biol Chem 1999; 274: 4743–4748. 54 Sepp-Lorenzino L, Rosen N. A farnesyl-protein transferase inhibitor induces p21 expression and G1 block in p53 wild type tumor cells. J Biol Chem 1998; 273: 20243–20251. 55 Karp JE, Lancet JE, Kaufmann SH, End DW, Wright JJ, Bol K et al. Clinical and biologic activity of the farnesyltransferase inhibitor R115777 in adults with refractory and relapsed acute leukemias: a phase 1 clinical–laboratory correlative trial. Blood 2001; 97: 3361–3369. 56 Fialkow PJ, Martin PJ, Najfeld V, Penfold GK, Jacobson RJ, Hansen JA. Evidence for a multistep pathogenesis of chronic myelogenous leukemia. Blood 1981; 58: 158–163. 57 Padua RA, McGlynn A, McGlynn H. Molecular, cytogenetic and genetic abnormalities in MDS and secondary AML. Cancer Treat Res 2001; 108: 111–157. 58 Gojo I, Karp JE. The impact of biology on the treatment of secondary AML. Cancer Treat Res 2001; 108: 231–255. 59 Kaizer H, Stuart RK, Brookmeyer R, Beschorner WE, Braine HG, Burns WH et al. Autologous bone marrow transplantation in acute leukemia: a phase I study of in vitro treatment of marrow with 4-hydroperoxycyclophosphamide to purge tumor cells. Blood 1985; 65: 1504–1510. 60 Borges-Walmsley MI, Walmsley AR. The structure and function of drug pumps. Trends Microbiol 2001; 9: 71–79. 61 Ban T. Pleiotropic, multidrug-resistant phenotype and P-glycoprotein: a review. Chemotherapy 1992; 38: 191–196. 62 Karp JE. MDR modulation in acute myelogenous leukemia: is it dead? Leukemia 2001; 15: 666–667. 63 Leith CP, Chen IM, Kopecky KJ, Appelbaum FR, Head DR, Godwin JE et al. Correlation of multidrug resistance (MDR1) protein expression with functional dye/drug efflux in acute myeloid leukemia by multiparameter flow cytometry: identification of discordant MDR/efflux+ and MDR1+/efflux cases. Blood 1995; 86: 2329–2342. 64 Borg AG, Burgess R, Green LM, Scheper RJ, Liu Yin JA. Overexpression of lung-resistance protein and increased Pglycoprotein function in acute myeloid leukaemia cells predict a poor response to chemotherapy and reduced patient survival. Br J Haematol 1998; 103: 1083–1091. 65 van den Heuvel-Eibrink MM, Sonneveld P, Pieters R. The prognostic significance of membrane transport-associated multidrug resistance (MDR) proteins in leukemia. Int J Clin Pharmacol Ther 2000; 38: 94–110. 66 Marie J-P. Drug resistance in hematologic malignancies. Curr Opin Oncol 2001; 13: 463–469. 67 Covelli A. Modulation of multidrug resistance (MDR) in hematological malignancies. Ann Oncol 1999; 10 (Suppl 6): 53–59. 68 Blair A, Hogge DE, Sutherland HJ. Most acute myeloid leukemia progenitor cells with long-term proliferative ability in vitro and in vivo have the phenotype CD34+/CD71/HLA-DR. Blood 1998; 92: 4325–4335. 69 Beck WT, Grogan TM, Willman CL, Cordon-Cardo C, Parham DM, Kuttesch JF et al. Methods to detect P-glycoproteinassociated multidrug resistance in patients’ tumors: consensus recommendations. Cancer Res 1996; 56: 3010–3020. 70 Lee EJ, George SL, Caligiuri M, Szatrowski TP, Powell BL, Lemke S et al. Parallel phase I studies of daunorubicin given with cytarabine and etoposide with or without the multidrug resistance modulator PSC-833 in previously untreated patients 60 years of Novel approaches to treat leukemia RA Larson et al 2379 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 age or older with acute myeloid leukemia: results of cancer and leukemia group B study 9420. J Clin Oncol 1999; 17: 2831– 2839. Baer MR, George SL, Dodge RK, O’Loughlin KL, Minderman H, Caligiuri MA et al. Phase 3 study of the multidrug resistance modulator PSC-833 in previously untreated patients 60 years of age and older with acute myeloid leukemia: Cancer and Leukemia Group B Study 9720. Blood 2002; 100: 1224–1232. Advani R, Saba HI, Tallman MS, Rowe JM, Wiernik PH, Ramek J et al. Treatment of refractory and relapsed acute myelogenous leukemia with combination chemotherapy plus the multidrug resistance modulator PSC 833 (Valspodar). Blood 1999; 93: 787–795. List AF, Kopecky KJ, Willman CL, Head DR, Persons DL, Slovak ML et al. Benefit of cyclosporine modulation of drug resistance in patients with poor-risk acute myeloid leukemia: a Southwest Oncology Group study. Blood 2001; 98: 3212–3220. Shepard RL, Cao J, Starling JJ, Dantzig AH. Modulation of Pglycoprotein but not MRP1- or BCRP-mediated drug resistance by LY335979. Int J Cancer 2003; 103: 121–125. Rubin EH, de Alwis DP, Pouliquen I, Green L, Marder P, Lin Y et al. A phase I trial of a potent P-glycoprotein inhibitor, Zosuquidar.3HCl trihydrochloride (LY335979), administered orally in combination with doxorubicin in patients with advanced malignancies. Clin Cancer Res 2002; 8: 3710–3717. Rowinsky EK, Smith L, Wang YM, Chaturvedi P, Villalona M, Campbell E et al. Phase I and pharmacokinetic study of paclitaxel in combination with biricodar, a novel agent that reverses multidrug resistance conferred by overexpression of both MDR1 and MRP. J Clin Oncol 1998; 16: 2964–2976. Peck RA, Hewett J, Harding MW, Wang YM, Chaturvedi PR, Bhatnagar A et al. Phase I and pharmacokinetic study of the novel MDR1 and MRP1 inhibitor biricodar administered alone and in combination with doxorubicin. J Clin Oncol 2001; 19: 3130–3141. Stewart A, Steiner J, Mellows G, Laguda B, Norris D, Bevan P. Phase I trial of XR9576 in healthy volunteers demonstrates modulation of P-glycoprotein in CD56+ lymphocytes after oral and intravenous administration. Clin Cancer Res 2000; 6: 4186–4191. Mistry P, Stewart AJ, Dangerfield W, Okiji S, Liddle C, Bootle D et al. In vitro and in vivo reversal of P-glycoprotein-mediated multidrug resistance by a novel potent modulator, XR9576. Cancer Res 2001; 61: 749–758. van Zuylen L, Sparreboom A, van der Gaast A, Nooter K, Eskens FALM, Brouwer E et al. Disposition of docetaxel in the presence of P-glycoprotein inhibition by intravenous administration of R101933. Eur J Cancer 2002; 38: 1090–1099. Javed A, Guo B, Hiebert S, Choi JY, Green J, Zhao SC et al. Groucho/TLE/R-esp proteins associate with the nuclear matrix and repress RUNX (CBF(alpha)/AML/PEBP2(alpha)) dependent activation of tissue-specific gene transcription. J Cell Sci 2000; 113: 2221–2231. Lutterbach B, Sun D, Schuetz J, Hiebert SW. The MYND motif is required for repression of basal transcription from the multidrug resistance 1 promoter by the t(8;21) fusion protein. Mol Cell Biol 1998; 18: 3604–3611. Dyer MJS, Oscier DG. The configuration of the immunoglobulin genes in B cell chronic lymphocytic leukemia. Leukemia 2002; 16: 973–984. Grever MR, Kopecky KJ, Coltman CA, Files JC, Greenberg BR, Hutton JJ et al. Fludarabine monophosphate: a potentially useful agent in chronic lymphocytic leukemia. Nouv Rev Fr Hematol 1988; 30: 457–459. Keating MJ, Kantarjian H, O’Brien S, Koller C, Talpaz M, Schachner J et al. Fludarabine: a new agent with marked cytoreductive activity in untreated chronic lymphocytic leukemia. J Clin Oncol 1991; 9: 44–49. Rai KR, Peterson BL, Appelbaum FR, Kolitz J, Elias L, Shepherd L et al. Fludarabine compared with chlorambucil as primary therapy for chronic lymphocytic leukemia. N Engl J Med 2000; 343: 1750–1757. Rai KR, Sawitsky A, Cronkite EP, Chanana AD, Levy RN, Pasternack BS. Clinical staging of chronic lymphocytic leukemia. Blood 1975; 46: 219–234. 88 Binet JL, Auquier A, Dighiero G, Chastang C, Piguet H, Goasguen J et al. A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer 1981; 48: 198–206. 89 Rai KR. Chronic lymphocytic leukaemia. Current strategy and new perspectives of treatment. Haematologica 1999; 84 (Suppl EHA-4): 94–95. 90 Stilgenbauer S, Bullinger L, Lichter P, Döhner H, for the German CLL Study Group (GCLLSG). Genetics of chronic lymphocytic leukemia: genomic aberrations and VHgene mutation status in pathogenesis and clinical course. Leukemia 2002; 16: 993–1007. 91 Dreger P, Brand R, Hansz J, Milligan D, Corradini P, Finke J et al. Treatment-related mortality and graft-versus-leukemia activity after allogeneic stem cell transplantation for chronic lymphocytic leukemia using intensity-reduced conditioning. Leukemia 2003; 17: 841–848. 92 Stilgenbauer S, Lichter P, Döhner H. Genetic features of B-cell chronic lymphocytic leukemia. Rev Clin Exp Hematol 2000; 4: 48–72. 93 Karnolsky IN. Cytogenetic abnormalities in chronic lymphocytic leukemia. Folia Med (Plovdiv) 2000; 42: 5–10. 94 Döhner H, Stilgenbauer S, Döhner K, Bentz M, Lichter P. Chromosome aberrations in B-cell chronic lymphocytic leukemia: reassessment based on molecular cytogenetic analysis. J Mol Med 1999; 77: 266–281. 95 Dearden C. Monoclonal antibody therapy of haematological malignancies. BioDrugs 2002; 16: 283–301. 96 Cheson BD. Hematologic malignancies: new developments and future treatments. Semin Oncol 2002; 29 (4 Suppl 13): 33–45. 97 Montserrat E. Rituximab in chronic lymphocytic leukemia. Semin Oncol 2003; 30 (1 Suppl 2): 34–39. 98 O’Brien SM, Kantarjian H, Thomas DA, Giles FJ, Freireich EJ, Cortes J et al. Rituximab dose-escalation trial in chronic lymphocytic leukemia. J Clin Oncol 2001; 19: 2165–2170. 99 Byrd JC, Murphy T, Howard RS, Lucas MS, Goodrich A, Park K et al. Rituximab using a thrice weekly dosing schedule in B-cell chronic lymphocytic leukemia and small lymphocytic lymphoma demonstrates clinical activity and acceptable toxicity. J Clin Oncol 2001; 19: 2153–2164. 100 Ravetch JV, Bolland S. IgG Fc receptors. Annu Rev Immunol 2001; 19: 275–290. 101 Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol 2001; 22: 633–640. 102 Clynes R, Takechi Y, Moroi Y, Houghton A, Ravetch JV. Fc receptors are required in passive and active immunity to melanoma. Proc Natl Acad Sci USA 1998; 95: 652–656. 103 Porcu P, Caligiuri MA. Cytokine–antibody combinations in the therapy of lymphoma. Biol Ther Lymphoma 2002; 5: 8–11. 104 Dancescu M, Rubio-Trujillo M, Biron G, Bron D, Delespesse G, Sarfati M. Interleukin 4 protects chronic lymphocytic leukemic B cells from death by apoptosis and upregulates Bcl-2 expression. J Exp Med 1992; 176: 1319–1326. 105 Foa R, Massaia M, Cardona S, Tos AG, Bianchi A, Attisano C et al. Production of tumor necrosis factor-alpha by B-cell chronic lymphocytic leukemia cells: a possible regulatory role of TNF in the progression of the disease. Blood 1990; 76: 393–400. 106 Podhorecka M, Dmoszynska A, Rolinski J, Wasik E. T type 1/type 2 subsets balance in B-cell chronic lymphocytic leukemia – the three-color flow cytometry analysis. Leuk Res 2002; 26: 657–660. 107 Vervoordeldonk SF, Merle PA, van Leeuwen EF, van der Schoot CE, von dem Borne AEGK, Slaper-Cortenbach ICM. Fcg receptor II (CD32) on malignant B cells influences modulation induced by anti-CD19 monoclonal antibody. Blood 1994; 83: 1632–1639. 108 Bannerji R, Kitada S, Flinn IW, Pearson M, Young D, Reed JC et al. Apoptotic-regulatory and complement-protecting protein expression in chronic lymphocytic leukemia: relationship to in vivo rituximab resistance. J Clin Oncol 2003; 21: 1466–1471. 109 Byrd JC, Kitada S, Flinn IW, Aron JL, Pearson M, Lucas D et al. The mechanism of tumor cell clearance by rituximab in vivo in patients with B-cell chronic lymphocytic leukemia: evidence of caspase activation and apoptosis induction. Blood 2002; 99: 1038–1043. 110 Byrd JC, Peterson BL, Morrison VA, Park K, Jacobson R, Hoke E et al. Randomized phase 2 study of fludarabine with concurrent Leukemia Novel approaches to treat leukemia RA Larson et al 2380 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 Leukemia versus sequential treatment with rituximab in symptomatic, untreated patients with B-cell chronic lymphocytic leukemia: results from Cancer and Leukemia Group B 9712 (CALGB 9712). Blood 2003; 101: 6–14. Reed JC, Kitada S, Kim Y, Byrd J. Modulating apoptosis pathways in low-grade B-cell malignancies using biological response modifiers. Semin Oncol 2002; 29: 10–24. Keating M, Hallek M. Alemtuzumab, the first monoclonal antibody (MAb) directed against CD52. Med Oncol 2002; 19 (Suppl): S1–S2. Domaga"a A, Kurpisz M. CD52 antigen: a review. Med Sci Monit 2001; 7: 325–331. Rai KR, Freter CE, Mercier RJ, Cooper MR, Mitchell BS, Stadtmauer EA et al. Alemtuzumab in previously treated chronic lymphocytic leukemia patients who also had received fludarabine. J Clin Oncol 2002; 20: 3891–3897. Keating MJ, Flinn I, Jain V, Binet J-L, Hillmen P, Byrd J et al. Therapeutic role of alemtuzumab (Campath-1H) in patients who have failed fludarabine: results of a large international study. Blood 2002; 99: 3554–3561. Kennedy B, Rawstron A, Carter C, Ryan M, Speed K, Lucas G et al. Campath-1H and fludarabine in combination are highly active in refractory chronic lymphocytic leukemia. Blood 2002; 99: 2245–2247. Mavromatis B, Cheson BD. Monoclonal antibody therapy of chronic lymphocytic leukemia. J Clin Oncol 2003; 21: 1874–1881. Shi JD, Bullock C, Hall WC, Wescott V, Wang H, Levitt DJ et al. In vivo pharmacodynamic effects of Hu1D10 (remitogen), a humanized antibody reactive against a polymorphic determinant of HLA-DR expressed on B cells. Leukemia Lymphoma 2002; 43: 1303–1312. Stockmeyer B, Schiller M, Repp R, Lorenz H-M, Kalden JR, Gramatzki M et al. Enhanced killing of B lymphoma cells by granulocyte colony-stimulating factor-primed effector cells and Hu1D10 – a humanized human leucocyte antigen DR antibody. Br J Haematol 2002; 118: 959–967. Pui C-H, Campana D, Evans WE. Childhood acute lymphoblastic leukaemia – current status and future perspectives. Lancet Oncol 2001; 2: 597–607. Pui C-H, Relling MV, Downing JR. Acute lymphoblastic leukemia. N Engl J Med (in press). Ferrando AA, Neuberg DS, Staunton J, Loh ML, Huard C, Raimondi SC et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell 2002; 1: 75–87. Pui C-H, Evans WE. Acute lymphoblastic leukemia. N Engl J Med 1998; 339: 605–615. Aricò M, Valsecchi MG, Camitta B, Schrappe M, Chessells J, Baruchel A et al. Outcome of treatment in children with Philadelphia chromosome-positive acute lymphoblastic leukemia. N Engl J Med 2000; 342: 998–1006. Pui C-H, Gaynon PS, Boyett JM, Chessells JM, Baruchel A, Kamps W et al. Outcome of treatment in childhood acute lymphoblastic leukaemia with rearrangements of the 11q23 chromosomal region. Lancet 2002; 359: 1909–1915. Pui C-H, Chessells JM, Camitta B, Baruchel A, Biondi A, Boyett JM et al. Clinical heterogeneity in childhood acute lymphoblastic leukemia with 11q23 rearrangements. Leukemia 2003; 17: 700–706. Yeoh EJ, Ross ME, Shurtleff SA, Williams WK, Patel D, Mahfouz R et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell 2002; 1: 133–143. Armstrong SA, Staunton JE, Silverman LB, Pieters R, den Boer ML, Minden MD et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet 2002; 30: 41–47. Armstrong SA, Kung AL, Mabon ME, Silverman LB, Stam RW, Den Boer ML et al. Inhibition of FLT3 in MLL. Validation of a therapeutic target identified by gene expression based classification. Cancer Cell 2003; 3: 173–183. Cheok MH, Yang W, Pui C-H, Downing JR, Cheng C, Naeve CW et al. Treatment-specific changes in gene expression discriminate in vivo drug response in human leukemia cells. Nat Genet 2003; 34: 85–90. 131 Pui C-H, Relling MV, Evans WE. Role of pharmacogenomics and pharmacodynamics in the treatment of acute lymphoblastic leukaemia. Best Pract Res Clin Haematol 2002; 15: 741–756. 132 Relling MV, Hancock ML, Rivera GK, Sandlund JT, Ribeiro RC, Krynetski EY et al. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst 1999; 91: 2001–2008. 133 Relling MV, Hancock ML, Boyett JM, Pui C-H, Evans WE. Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood 1999; 93: 2817–2823. 134 Pui C-H, Relling MV. Topoisomerase II inhibitor-related acute myeloid leukaemia. Br J Haematol 2000; 109: 13–23. 135 Relling MV, Rubnitz JE, Rivera GK, Boyett JM, Hancock ML, Felix CA et al. High incidence of secondary brain tumors after radiotherapy and antimetabolites. Lancet 1999; 354: 34–39. 136 Thomsen JB, Schrøder H, Kristinsson J, Madsen B, Szumlanski C, Weinshilboum R et al. Possible carcinogenic effect of 6mercaptopurine on bone marrow stem cells: relation to thiopurine metabolism. Cancer 1999; 86: 1080–1086. 137 Krajinovic M, Costea I, Chiasson S. Polymorphism of the thymidylate synthase gene and outcome of acute lymphoblastic leukaemia. Lancet 2002; 359: 1033–1034. 138 Urano W, Taniguchi A, Yamanaka H, Tanaka E, Nakajima H, Matsuda Y et al. Polymorphisms in the methylenetetrahydrofolate reductase gene were associated with both the efficacy and the toxicity of methotrexate used for the treatment of rheumatoid arthritis, as evidenced by single locus and haplotype analyses. Pharmacogenetics 2002; 12: 183–190. 139 Ulrich CM, Yasui Y, Storb R, Schubert MM, Wagner JL, Bigler J et al. Pharmacogenetics of methotrexate: toxicity among marrow transplantation patients varies with the methylenetetrahydrofolate reductase C677T polymorphism. Blood 2001; 98: 231–234. 140 Taub JW, Matherly LH, Ravindranath Y, Kaspers G-JL, Rots MG, Zantwijk CH. Polymorphisms in methylenetetrahydrofolate reductase and methotrexate sensitivity in childhood acute lymphoblastic leukemia. Leukemia 2002; 16: 764–765. 141 Pui C-H, Campana D. New definition of remission of childhood acute lymphoblastic leukemia. Leukemia 2000; 14: 783–785. 142 Szczepanski T, Orfão A, van der Velden VJH, San Miguel JF, van Dongen JJM. Minimal residual disease in leukaemia patients. Lancet Oncol 2001; 2: 409–417. 143 Coustan-Smith E, Sancho J, Hancock ML, Boyett JM, Behm FG, Raimondi SC et al. Clinical importance of minimal residual disease in childhood acute lymphoblastic leukemia. Blood 2000; 96: 2691–2696. 144 Panzer-Grümayer ER, Schneider M, Panzer S, Fasching K, Gadner H. Rapid molecular response during early induction chemotherapy predicts a good outcome in childhood acute lymphoblastic leukemia. Blood 2002; 95: 790–794. 145 Coustan-Smith E, Sancho J, Behm FG, Hancock ML, Razzouk RI, Ribeiro RC et al. Prognostic significance of measuring early clearance of leukemic cells by flow cytometry in childhood acute lymphoblastic leukemia. Blood 2002; 100: 52–58. 146 Neale GAM, Coustan-Smith E, Pan Q, Chen X, Gruhn B, Stow P et al. Tandem application of flow cytometry and polymerase chain reaction for comprehensive detection of minimal residual disease in childhood acute lymphoblastic leukemia. Leukemia 1999; 13: 1221–1226. 147 Coustan-Smith E, Sancho J, Hancock ML, Razzouk BI, Ribeiro RC, Rivera GK et al. Use of peripheral blood instead of bone marrow to monitor residual disease in children with acute lymphoblastic leukemia. Blood 2002; 100: 2399–2402. 148 van der Velden VHJ, Jacobs DCH, Wijkhuijs AJM, Comans-Bitter WM, Willemse MJ, Hählen K et al. Minimal residual disease levels in bone marrow and peripheral blood are comparable in children with T cell acute lymphoblastic leukemia (ALL), but not in precursor-B-ALL. Leukemia 2002; 16: 1432–1436. 149 Pui C-H, Cheng C, Leung W, Rai SN, Rivera GK, Sandlund JT et al. Extended follow-up of long-term survivors of childhood acute lymphoblastic leukemia. N Engl J Med 2003; 349: 640– 649. 150 Vilmer E, Suciu S, Ferster A, Bertrand Y, Cavé H, Thyss A et al. Long-term results of three randomized trials (58831, 58832, 58881) in childhood acute lymphoblastic leukemia: a CLCG- Novel approaches to treat leukemia RA Larson et al 2381 151 152 153 154 155 156 157 158 159 160 161 162 163 164 165 166 167 168 169 170 EORTC report. Children Leukemia Cooperative Group. Leukemia 2000; 14: 2257–2266. Manera R, Ramirez I, Mullins J, Pinkel D. Pilot studies of speciesspecific chemotherapy of childhood acute lymphoblastic leukemia using genotype and immunophenotype. Leukemia 2000; 14: 1354–1361. Pui C-H. Toward optimal central nervous system-directed treatment in childhood acute lymphoblastic leukemia. J Clin Oncol 2003; 21: 179–181. Mahmoud HH, Rivera GK, Hancock ML, Krance RA, Kun LE, Behm FG et al. Low leukocyte counts with blast cells in cerebrospinal fluid of children with newly diagnosed acute lymphoblastic leukemia. N Engl J Med 1993; 329: 314–319. Bürger B, Zimmermann M, Mann G, Kühl J, Löning L, Riehm H et al. Diagnostic cerebrospinal fluid examination in children with acute lymphoblastic leukemia: significance of low leukocyte counts with blasts or traumatic lumbar puncture. J Clin Oncol 2003; 21: 184–188. Gajjar A, Harrison PL, Sandlund JT, Rivera GK, Ribeiro RC, Rubnitz JE et al. Traumatic lumbar puncture at diagnosis adversely affects outcome in childhood acute lymphoblastic leukemia. Blood 2000; 96: 3381–3384. Howard SC, Gajjar AJ, Cheng C, Kritchevsky SB, Somes GW, Harrison PL et al. Risk factors for traumatic and bloody lumbar puncture in children with acute lymphoblastic leukemia. JAMA 2002; 288: 2001–2007. Gandhi V, Plunkett W, Rodriguez Jr CO, Nowak BJ, Du M, Ayres M et al. Compound GW506U78 in refractory hematologic malignancies: relationship between cellular pharmacokinetics and clinical response. J Clin Oncol 1998; 16: 3607–3615. Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med 2001; 344: 1038–1042. Kantarjian HM, Gandhi V, Kozuch P, Faderl S, Giles F, Cortes J et al. Phase I clinical and pharmacology study of clofarabine in patients with solid and hematologic cancers. J Clin Oncol 2003; 21: 1167–1173. Navolanic PM, Pui C-H, Larson RA, Bishop MR, Pearce TE, Cairo MS et al. Elitekt-rasburicase: an effective means to prevent and treat hyperuricemia associated with tumor lysis syndrome: a Meeting Report, Dallas, Texas, January 2002. Leukemia 2003; 17: 499–514. Hande KR, Hixson CV, Chabner BA. Postchemotherapy purine excretion in lymphoma patients receiving allopurinol. Cancer Res 1981; 41: 2273–2279. Cunningham SG. Fluid and electrolyte disturbances associated with cancer and its treatment. Nurs Clin N Am 1982; 17: 579–593. Boles J-M, Dutel J-L, Briere J, Mialon P, Robasckiewicz M, Garre M et al. Acute renal failure caused by extreme hyperphosphatemia after chemotherapy of an acute lymphoblastic leukemia. Cancer 1984; 53: 2425–2429. Stapleton FB, Strother DR, Roy III S, Wyatt RJ, McKay CP, Murphy SB. Acute renal failure at onset of therapy for advanced stage Burkitt lymphoma and B cell acute lymphoblastic lymphoma. Pediatrics 1988; 82: 863–869. Fleming DR, Doukas MA. Acute tumor lysis syndrome in hematologic malignancies. Leukemia Lymphoma 1992; 8: 315–318. Stokes DN. The tumour lysis syndrome: intensive care aspects of paediatric oncology. Anaesthesia 1989; 44: 133–136. Hande KR, Garrow GC. Acute tumor lysis syndrome in patients with high-grade non-Hodgkin’s lymphoma. Am J Med 1993; 94: 133–139. Stucky LA. Acute tumor lysis syndrome: assessment and nursing implications. Oncol Nurs Forum 1993; 20: 49–59. Chasty RC, Liu-Yin JA. Acute tumour lysis syndrome. Br J Hosp Med 1993; 49: 488–492. Dietz KA, Flaherty AM. Oncologic emergencies. In: Groenwald SL, Frogge MH, Goodman M, Yarbro CH (eds). Cancer Nursing: Principles and Practice 3rd edn. Boston: Jones & Bartlett, 1993, pp 821–824. 171 Kedar A, Grow W, Neiberger RE. Clinical versus laboratory tumor lysis syndrome in children with acute leukemia. Pediatr Hematol Oncol 1995; 12: 129–134. 172 Lawrence J. Critical care issues in the patient with hematologic malignancy. Semin Oncol Nurs 1994; 10: 198–207. 173 Veenstra J, Krediet RT, Somers R, Arisz L. Tumour lysis syndrome and acute renal failure in Burkitt’s lymphoma. Description of 2 cases and a review of the literature on prevention and management. Neth J Med 1994; 45: 211–216. 174 Lorigan PC, Woodings PL, Morgenstern GR, Scarffe JH. Tumour lysis syndrome, case report and review of the literature. Ann Oncol 1996; 7: 631–636. 175 Jones DP, Mahmoud H, Chesney RW. Tumor lysis syndrome: pathogenesis and management. Pediatr Nephrol 1995; 9: 206–212. 176 Kjellstrand CM, Campbell II DC, von Hartitzsch B, Buselmeier TJ. Hyperuricemic acute renal failure. Arch Intern Med 1974; 133: 349–359. 177 Mahmoud HH, Leverger G, Patte C, Harvey E, Lascombes F. Advances in the management of malignancy-associated hyperuricaemia. Br J Cancer 1998; 77 (Suppl 4): 18–20. 178 Ten Harkel ADJ, Kist-Van Holthe JE, Van Weel M, Van der Vorst MMJ. Alkalinization and the tumor lysis syndrome. Med Pediatr Oncol 1998; 31: 27–28. 179 Wolf G, Hegewisch-Becker S, Hossfeld DK, Stahl RAK. Hyperuricemia and renal insufficiency associated with malignant disease: urate oxidase as an efficient therapy? Am J Kidney Dis 1999; 34: E20. 180 Dillman RO. Infusion reactions associated with the therapeutic use of monoclonal antibodies in the treatment of malignancy. Cancer Metastasis Rev 1999; 18: 465–471. 181 Bishop MR, Coccia PF. Tumor lysis syndrome. In: Abeloff MD, Armitage JO, Lichter AS, Niederhuber JE (eds). Clinical Oncology 2nd edn. New York: Churchill Livingstone, 2000, pp 750–754. 182 Flombaum CD. Metabolic emergencies in the cancer patient. Semin Oncol 2000; 27: 322–334. 183 Arrambide K, Toto RD. Tumor lysis syndrome. Semin Nephrol 1993; 13: 273–280. 184 Ezzone SA. Tumor lysis syndrome. Semin Oncol Nurs 1999; 15: 202–208. 185 Jeha S. Tumor lysis syndrome. Semin Hematol 2001; 38 (4 Suppl 10): 4–8. 186 Hogan DK, Rosenthal LD. Oncologic emergencies in the patient with lymphoma. Semin Oncol Nurs 1998; 14: 312–320. 187 Kelly KM, Lange B. Oncologic emergencies. Pediatr Clin N Am 1997; 44: 809–830. 188 Easton J, Noble S, Jarvis B. Rasburicase. Paediatr Drugs 2001; 3: 433–437. 189 Laboureur P, Langlois C. Urate-oxydase d’Aspergillus flavus: I. Obtention, purification, propriétés. Bull Soc Chim Biol (Paris) 1968; 50: 811–825. 190 Chanteclair G, Cartault F, Humbert J-C, Olive D, Neimann N. Place de l’urate-oxydase dans la prévention de l’hyperuricémie en hématologie infantile. Rev Int Pediatr 1974-1975; 50: 47–56. 191 Leplatois P, Le Douarin B, Loison G. High-level production of a peroxisomal enzyme: Aspergillus flavus uricase accumulates intracellularly and is active in Saccharomyces cerevisiae. Gene 1992; 122: 139–145. 192 Pui C-H, Relling MV, Lascombes F, Harrison PL, Struxiano A, Mondesir J-M et al. Urate oxidase in prevention and treatment of hyperuricemia associated with lymphoid malignancies. Leukemia 1997; 11: 1813–1816. 193 Leach M, Parsons RM, Reilly JT, Winfield DA. Efficacy of urate oxidase (uricozyme) in tumour lysis induced urate nephropathy. Clin Lab Haematol 1998; 20: 169–172. 194 Fam AG. Difficult gout and new approaches for control of hyperuricemia in the allopurinol-allergic patient. Curr Rheumatol Rep 2001; 3: 29–35. 195 Goldman SC, Holcenberg JS, Finklestein JZ, Hutchinson R, Kreissman S, Johnson FL et al. A randomized comparison between rasburicase and allopurinol in children with lymphoma or leukemia at high risk for tumor lysis. Blood 2001; 97: 2998–3003. 196 Pui C-H. Introduction – optimal treatment of malignancies associated with hyperuricemia. Semin Hematol 2001; 38 (4 Suppl 10): 1–3. Leukemia Novel approaches to treat leukemia RA Larson et al 2382 197 Patte C, Sakiroglu O, Sommelet D. European experience in the treatment of hyperuricemia. Semin Hematol 2001; 38 (4 Suppl 10): 9–12. 198 Pui C-H. Urate oxidase in the prophylaxis or treatment of hyperuricemia: the United States experience. Semin Hematol 2001; 38 (4 Suppl 10): 13–21. 199 Pui C-H, Jeha S, Irwin D, Camitta B. Recombinant urate oxidase (rasburicase) in the prevention and treatment of malignancy- Leukemia associated hyperuricemia in pediatric and adult patients: results of a compassionate-use trial. Leukemia 2001; 15: 1505–1509. 200 Pui C-H, Mahmoud HH, Wiley JM, Woods GM, Leverger G, Camitta B et al. Recombinant urate oxidase for the prophylaxis or treatment of hyperuricemia in patients with leukemia or lymphoma. J Clin Oncol 2001; 19: 697–704. 201 Pui C-H. Rasburicase: a potent uricolytic agent. Expert Opin Pharmacother 2002; 3: 433–452.