Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Enzyme Kinetics
Introduction to Enzymes
• Drive all the processes in a living cell
Proteins or RNA
• Most enzymes are proteins and some are RNA (ribozymes).
Catalyst
• Enzymes are not consumed in the reaction. They participate in the reaction
but regenerate at the end of the reaction.
Greater reaction specificity
• Enzymatic reactions are highly specific. The degree of enzyme specificity
varies and dependent on the enzyme.
Higher reaction rate
• The rates of enzyme-catalyzed reactions are 106 to 1012 times faster than
uncatalyzed reactions.
Milder reaction conditions
• Enzyme catalyzed reactions occur at close to physiological temperatures and
pH.
Capacity for regulation
• Enzymatic reactions can be regulated by substrate, product, and enzyme
concentration and also by allosteric and covalent modification of enzymes.
1
Enzyme Specificity
Substrate binding site and active site
• Active site is a pocket lined with amino acids (catalytic amino acids) precisely
positioned to participate in binding and catalysis of specific substrates.
Fischer’s lock and key concept of enzyme action
• Fischer proposed that “… an enzyme and substrate must fit together like a
lock and key.”
• This model provides a limited and rigid view of enzymes. Inefficient catalysis
of small ligands and effect of allosteric ligands cannot be explained.
Induced fit model
• The substrate initially forms a relatively
weak complex with the enzyme.
• Substrate binding induces a
conformational change in the enzyme that
strengthens substrate binding as well as
brings the catalytic groups close to the
substrate.
substrate
+
induced fit
ES complex
enzyme
Evidence:
• X-ray crystal structures of hexokinase with and without glucose. The
hexokinase closes around glucose to bring the catalytic groups in close
proximity to glucose.
ATP + glucose
ATP + H2O
ADP + glucose-6-phosphate
ADP + phosphate
In the cell, the concentration of H2O is much higher
than glucose
2
Binding of glucose induces a large conformational change
The two lobes swing together by 12 A
Glucose is completely engulfed by the protein
Rate acceleration
• Enzymes accelerate rates about 106 to 1014 relative to uncatalyzed reactions.
E.g., Hydrolysis of urea occurs nonenzymatically at a rate of 4 x 10-5 s-1 at 100
o
C (half life of 4.8 hours) and 3 x 10-10 s-1 at 20 oC (half life of 73 years!). The
urease catalyzed reaction occurs at a rate of 3 x 104 s-1 20 oC (half life of 23
microseconds) which represents close to 1014-fold acceleration in rate.
Activation energy and Transition State
• All chemical reactions have an energy barrier, the activation energy barrier
(∆G‡) that must be overcome for reaction to occur. The energy barrier can
be overcome by increasing the temperature, an option not possible for living
organisms. Enzymes employ other techniques.
• Enzyme increase rate by stabilizing the transition state
• At the peak of the energy barrier is the activated complex known as the
transition state.
• In the transition state structure, bonds are being made or broken or more
strained and in improbable states, and therefore are energetically
unfavorable.
• The life time of transition state complex is very short (it is transient) and the
complex cannot be isolated.
3
uncatalyzed reaction
Fre ∆ ‡
G
e
ene
rgy
Enzyme-catalyzed reaction
transition state
∆G‡
∆G
progress of reaction
∆G
progress of reaction
4
Kinetic Concepts
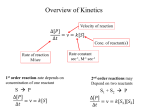
Velocity
Speed of the reaction or rate of the reaction.
Measure changes in concentration of reactant (or product) as a function of time.
Rate can be described either by the appearance of products or the
disappearance of reactants.
A → B
v = -d[A]/dt = d[B]/dt
[B]
slope = v
time
In an enzymatic reaction, velocity decreases at
longer times when product formation levels off.
The most common reasons for this kinetic
behavior is the depletion of substrate and
increase in reverse reaction and product
inhibition. The other reason could be enzyme
inactivation due to denaturation.
[P]
time
It is therefore important to determine the velocity at initial times –initial velocity.
The rule is to measure the velocity during the period when about 10% of the
substrate (S) is converted to product (P).
5
Enzyme assays
Monitor change in concentration of substrate or product with time
Change in some physical or chemical property such as absorbance,
fluorescence, or pH.
It is generally advisable to measure the property of the product or product
formation rather than utilization of substrate for accurate measurements.
First order reaction
Consider an irreversible reaction:
A → B
The rate of a first order reaction is proportional to the concentration of a single
species:
v = k[A]
-d[A]/dt = k[A]
where k= rate constant. If the concentration of the reactant is expressed in molar
scale, then the units of this proportionality constant (k) for a first order reaction is
time-1.
d[A]/dt = -k[A]
d[A]/[A] = -kdt
upon integration,
ln[A] = -kt + C
t=0, [A] = [Ao] therefore C = ln[Ao]
ln [A] = -kt + ln[Ao]
[A] = [Ao]e-kt
… this is an exponential equation.
6
Ao
ln[Ao]
[A]
slope = -k
ln[A]
[Ao]/2
t1/2
time
time
log[Ao]
t1/2 = 0.693/k
slope = -k/2.303
log[A]
time
The practical advantage of first-order rate constants arises from it being
independent of the concentration of reactant. So the rate constant can be
obtained from the half-time for the appearance or disappearance of a product or
reactant without knowing its absolute concentration.
Now, consider a reversible reaction:
k1
A
B
k-1
Here A does not completely transform into B. At the end of the reaction (that is,
at equilibrium) there is an equilibrium concentration of A.
Keq = [B]/[A] = k1/k-1
dA/dt = -k1[A] + k-1[B]
d[A]/dt = -k1[A] + k-1 {[Ao] - [A]}
Upon integration (for details see page 130 of Fersht)
[ At ] =
[ Ao ]k1
exp[− ( k1 + k − 1 )t ] + k − 1
k1 + k − 1
7
A is converted to B with an observed exponential rate constant equal to k1 + k-1.
The amplitude of the exponential is k1/k1+k-1
100%
[A]
[A]eq
time
Second order reaction
A+A→ B
-d[A]/dt = k[A]2
-d[A]/[A]2 = kdt
upon integration,
1/[A] = kt + C when t=0, [A]=[Ao] therefore C=1/[Ao]
1/[A] = kt + 1/[Ao]
k
1/[A]
1/[Ao]
time
8
The rate constant k for a second order reaction has the units of M-1time-1. The
second order rate constant is therefore dependent on the concentration of
reactant.
Pseudo first-order rate constants
A+B→ P
v = k[A][B]
if [A]>>[B], then concentration of A does not change during the course of the
reaction, therefore [A] is approximately a constant.
v = k’[B], where k’= k[A]
k’is referred to as the psuedo first-order rate constant.
The above is the most generally useful kinetic method for the elucidation of
reaction mechanism.
Order of the reaction
The order of the reaction describes the number of molecules to which the
reaction rate is proportional, and it does not necessarily indicate the number of
molecules involved in the reaction.
To determine the order of the reaction, one follows the rate of the reaction as
function of reactant concentration.
If, plot of [A] versus time is linear -zero order reaction
If, ln[A] versus time is linear
-first order reaction
If, 1/[A] versus time is linear
-second order reaction
9
Enzyme Kinetics
The purified enzyme is characterized by measuring its steady state kinetic
parameters –Km and Vmax.
A simple one substrate reaction is shown below
Substrate (S)
Product (P)
When this reaction is catalyzed by an enzyme E, the enzyme forms a reversible
complex with the substrate (ES) which is then converted to P.
The rate constant for formation of ES is k1 and the rate constant for dissociation
of the ES complex is k-1. The conversion of ES to E + P occurs with a rate
constant k2 and the reverse reaction rate is k-2.
k1
E.S
E+S
k2
E+P
k-2
k-1
The rate of formation of P is the velocity of the reaction at any time t. If the initial
velocity is measured then the [P] is close to zero and therefore the rate of the
reverse reaction will be negligible and the above equation simplifies to
k1
E.S
E+S
k2
E+P
k-1
There are two ways to derive kinetic equations for describing the above reaction.
1) By assuming that ES is in rapid equilibrium with E and S (Michaelis-Menten
derivation).
2) By assuming a steady-state condition; that is, the rate of formation of ES
equals the rate of its breakdown (Briggs-Haldane derivation).
10
Michaelis-Menten derivation
Assumptions:
1) ES is in rapid equilibrium with free E and S.
2) Formation of product is proportional to [ES]
3) [S] >> [E] so change in [S] is negligible.
Ks
E+S
ES
k2
E+P
[ E ][ S ]
[ ES ]
[ Et ] = [ E ] + [ ES ]
([ Et ] − [ ES ])[ S ]
Ks =
[ ES ]
[ E ][ S ]
[ ES ] =
K s + [S ]
Ks =
v = k 2 [ ES ] = k cat [ ES ] =
k cat [ E ][ S ]
K s + [S ]
Vmax = k cat [ E ]
v=
V max [ S ]
K s + [S ]
The above is the Michaelis-Menten equation. The velocity (v) versus [S] is a
hyperbola.
11
Briggs-Haldane derivation
Most enzymes do not obey Michaelis-Menten kinetics. That is, the ES is not in
rapid equilibrium with free E and S.
k1
E.S
E+S
k2
E+P
k-1
In reality, k-1 is not much greater than k2. In such a case, the steady-state
assumption is used in deriving an equation for the above reaction.
Assumptions
1) ES is in a steady-state. That is, the rate of ES formation is equal to its
breakdown.
2) The rate of product formation is proportional to [ES].
3) [S]>>[E] so [S] remains constant during the measurement.
Steady state assumption states that d[ES]/dt = 0
The rate of formation of ES = k1[E][S]
The rate of utilization of ES = k-1[ES]+k2[ES] = (k-1+k2)[ES]
k1[E][S] =(k-1+k2)[ES]
[Et] = [E]+[ES] or [E] = [Et]-[ES]
k1([Et]-[ES])[S] =(k-1+k2)[ES]
[Et][S]-[ES][S] ={(k-1+k2)/ k1 }[ES]
k-1+k2)/ k1 = Km
[ES] = [Et][S]/(Km + [S])
v= k2[ES] and Vmax = k2[Et]
v = Vmax [S]/(Km + [S])
12
Determination of Vmax and Km
1
Km
1
=
+
v Vmax[S] Vmax
and 1/v is plotted versus 1/[S] which is a
straight line. The y-intercept corresponds to
1/Vmax and the x-intercept is equal to -1/Km.
This is known as the Lineweaver-Burk plot or
the double reciprocal plot.
1
vo
Enzyme 2
Vmax
2
0
0
[Substrate]
Km1 Km2
Large Km of enzyme 2
reflects a low affinity
of enzyme for the
substrate.
Small Km for enzyme 1
reflects a high affinity
of enzyme forsubstrate.
the
substrate.
1
Vmax
1
Km
Enzyme 1
Vmax
Reaction velocity (vo)
When the reaction velocity is plotted versus
[S], a hyperbolic dependence is observed
which can be fit to the above Michaelis Menten
equation to obtain Vmax and Km. Alternatively,
the above equation can be modified as follows:
1
[S]
Effect of substrate concentration on
reaction velocities for two enzymes,
enzyme 1 with a small Km, enzyme 2
Lineweaver-Burke plot.
with a large Km.
13
Are Km and Ks identical?
They are not identical.
Meaning of Ks
Ks is a true equilibrium dissociation constant. It is the same as the Kd of [ES].
Meaning of Km
The Km is the concentration of substrate at which Vmax is half. The units of Km
are molar (M). The value of Km is independent of enzyme concentration or its
purity but depends on pH, temperature, and presence of inhibitors or activators,
ionic strength.
Meaning of Vmax and kcat
The maximum velocity with which an enzyme catalyzes the conversion of S to P.
Vmax is linearly dependent on enzyme concentration: Vmax = kcat.[E]. Thus,
enzyme concentration can directly regulate the velocity of a reaction.
kcat is the turnover number of an enzyme reflecting the number of mols of
substrates converted to products per sec per mol of enzyme.
kcat is a constant for an enzyme and substrate, similar to Km, and changes only
with pH and temperature.
Km is often misinterpretated as the dissociation constant and kcat as the rate of
the chemical reaction.
Km may be considered an apparent dissociation constant. K m =
where ∑ [ES] is the sum of all bound enzyme species.
[ E ][S ]
∑ [ES ]
kcat is the first order rate constant for conversion of ES to EP. kcat is a function of
all the microscopic rate constants and it cannot be assigned to any particular
step in the reaction.
For example, for a minimal enzymatic mechanism:
E+S
k cat =
ES
k 2 k3
k 2 + k − 1 + k3
EP
E+P
and K m =
k 2 k3 + k − 1k − 2 + k − 1k3
k1 ( k 2 + k − 2 + k3 )
14
Both of which are complicated functions of each of intrinsic or microscopic rate
constants.
kcat/Km
Often referred to as the specificity constant. It is a useful term when comparing
the efficiency of catalysis of several substrates by the same enzyme or
comparing the efficiencies of several mutant enzymes.
E.g., following are the kcat and Km values for the hydrolysis of substrates by
trypsin. Which one is a better substrate?
Substrate
1
2
kcat
24 s-1
99 s-1
Km
1.5 uM
5.8 uM
Kcat/Km
16 uM-1s-1
17 uM-1s-1
What we have learned from steady state kinetic experiments:
The value of kcat provides the lower limit for the enzymatic conversion of
substrate to product.
kcat/Km provides the lower limit on the apparent second order rate constant for
substrate binding.
The order of substrate binding and product release can be defined.
15
Presteady state enzyme kinetics
The kinetic analysis of an enzyme mechanism begins with the analysis in steady
state. The kinetic mechanism derived from steady state kinetics allows one to
specify the order of addition of substrates and order of release of products.
However, steps in the reaction are not defined or directly measured. Direct
measurements of reactions at the enzyme active site are necessary to establish
the kinetic mechanism. One can follow the formation and decay of enzyme
species or intermediates directly to obtain the microscopic rate constants.
To measure the steady-state kinetics of a reaction, the [S]>>[E] and one
measures product from multiple turnovers of E.
In a presteady state experiment, one measure the kinetics of product formation
in the first turnover. This provides information about steps from substrate
E+S
ES
EI
EP
E+P
binding to product release.
The goal of a complete kinetic analysis is to determine the kinetic rate constants
of each step in the pathway. This knowledge provides the free energy profile of
the reaction and allows one to understand the relationship between binding
energy and catalytic efficiency. It provides the basis to study structure-function
by determining the effect of amino acid mutations.
Direct measurement of microscopic kinetic rate constants and
enzyme intermediates:
To observe and measure the kinetics of enzyme intermediates, rapid kinetic
techniques are employed.
Stopped-flow methods.
Rapid chemical quench-flow methods.
16
Stopped-flow Apparatus
enzyme
Light
substrate
Absorbance changes
stop syringe
Light scattering or Fluorescence changes
Stopped-flow method is used to measure the kinetics of reaction when there is
an optical change during the reaction. For example, absorbance changes in
enzymes containing heme, pyridoxal phosphate, NADH, and flavins cofactors.
Similarly, fluorescence changes of tyrosine and tryptophan residues in proteins
can be measured.
17
If there is no optical change during the reaction then stopped-flow techniques
cannot be used. One can then use radiolabeled substrates and the chemical
quench-flow apparatus to measure the rapid kinetics of the enzymatic reaction.
Rapid Chemical Quench-flow Apparatus
enzyme
substrate
chemical quench
reaction delay
line
In the above apparatus, one varies the length of the reaction delay line to vary
the reaction time.
Mixing needs to be rapid in order to maintain turbulent flow.
Common quenchers: 1N HCl, 1N NaOH, EDTA, SDS -to stop the reaction
rapidly.
18
Kinetics of substrate binding
One-step binding
k
E + S 1 → ES
One can measure the kinetics of substrate or ligand binding to the protein using
the stopped-flow technique and monitoring the change in some optical property.
For example, in many instances, substrate or ligand binding to the enzyme
results in change in the intrinsic fluorescence of the protein (fluorescence of
tyrosine or tryptophan) due to energy transfer or conformational changes in the
protein accompanying substrate binding.
The ligand may be fluorescent and upon binding its fluorescence or absorbance
properties change.
k
E + S 1 → ES
goal is the measure k1.
d(ES)/dt = k1 [E] [S]
If [S]>>[E] , then [S] remains constant over the time of the reaction
and, d(ES)/dt = k1’[E] where k1’= k1[S]
Upon integration,
[ES] = [Eo](1-e-k1[S]t)
Thus, free enzyme disappears and ES is formed in an exponential manner with
time, and the rate constant of the exponential = k1[S]
19
k1 M-1 s-1.
kobs
(s-1)
fluorescence
[S] M
kobs = 0.693/t1/2
t1/2
time
kobs = k1[S]
Therefore plot of kobs versus [S] should be linear with slope = k1.
20
Most binding reactions are however reversible
k1
E+S
ES
k-1
If [S] >> [E], then the [S] does not change much over time
k1[S]
therefore, E
ES
k-1
[ ES ]
K1[S ]
=
[ Eo ] 1 + K1 [ S ]
where K1 is the equilibrium association constant =
[ ES ]
[ E ][S ]
[ ES ]
K1 [ S ]
− k t
=
(1 − e obs )
[ Eo] 1 + K1[S ]
kobs = k1[S] + k-1 (this is an equation for a line with an intercept)
[S]
k1 M-1 s-1.
kobs
(s-1)
[ES]
k-1
time
[S] M
21
Presteady state burst kinetics
In a presteady state experiment, when the enzyme is first mixed with substrate
(under conditions [S]>[E]) one can observe a burst of product formation at a rate
faster than steady state turnover. This presteady state burst is due to the
accumulation of product at the active site of enzyme. On quenching the reaction
mixture with a denaturing agent, the enzyme-bound product is released. The
total product is therefore the sum of product free in solution at the time of
addition of quench and the enzyme-bound product which is released at the time
of addition of quench.
k
k3
ES 2 → EP → E + P
[ P ] [ EP] + [ P]
=
= Ao(1 − e − λt ) + k cat t
[ Eo ]
[ Eo ]
steady state phase
[P]+[EP]
Ao
presteady state burst phase
time
λ= k 2 + k3
Ao = (
k2
)2
k 2 + k3
k cat =
k 2 k3
k 2 + k3
22
The amplitude Ao will be 1 per enzyme site if the formation of product (k2) is
much greater than the release of product (k3). k2>>k3
If there is no burst, then either the chemical reaction (k2) or a step preceding it
(such as substrate binding) is rate limiting. k3>>k2
Using presteady state experiments it is possible to dissect the kinetic
mechanism into its microscopic rate constants. The advantage of presteady
state kinetics is its ability to directly observe reaction intermediates.
23
24