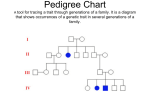
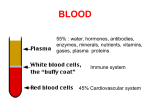
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Hemophilia And Von Willebrand Disease In Children: Emergency Department Evaluation And Management Abstract Adam E. Vella, MD, FAAP Associate Professor of Emergency Medicine, Pediatrics, and Medical Education, Director Of Pediatric Emergency Medicine, Icahn School of Medicine at Mount Sinai, New York, NY Associate Editor-in-Chief Vincent J. Wang, MD, MHA Associate Professor of Pediatrics, Keck School of Medicine of USC; Associate Division Head, Division of Emergency Medicine, Children's Hospital Los Angeles, Los Angeles, CA Editorial Board Jeffrey R. Avner, MD, FAAP Professor of Clinical Pediatrics and Chief of Pediatric Emergency Medicine, Albert Einstein College of Medicine, Children’s Hospital at Montefiore, Bronx, NY Steven Bin, MD Associate Clinical Professor, Division of Pediatric Emergency Medicine, UCSF Benioff Children’s Hospital, University of California, San Francisco, CA Richard M. Cantor, MD, FAAP, FACEP Professor of Emergency Medicine and Pediatrics, Director, Pediatric Emergency Department, Medical Director, Central New York Poison Control Center, Golisano Children's Hospital, Syracuse, NY Volume 12, Number 9 Authors Kevin R. Schwartz, MD Instructor of Pediatrics, Harvard Medical School, Department of Pediatrics and Department of Emergency Medicine, Massachusetts General Hospital, Boston, MA Max Rubinstein, MD Department of Pediatrics, Massachusetts General Hospital, Harvard Medical School, Boston, MA Peer Reviewers Hemophilia and von Willebrand disease are the most common inherited bleeding disorders encountered in the emergency department. Evidence suggests that the management of bleeding disorders in the emergency department is currently suboptimal, and literature to guide evaluation and management in this setting is limited, though some guidelines do exist. The emergency clinician must have a high index of suspicion for new diagnoses, particularly in young patients with unprovoked bleeding and children with multiple or severe bleeds. The foundation of hemophilia treatment is urgent clotting factor replacement, with replacement goals guided by the presenting complaint. Bleeding in von Willebrand disease may be treated with products containing von Willebrand factor or with desmopressin. This review focuses on the epidemiology, pathophysiology, common presentations, evaluation strategies, and emergency management of these bleeding disorders. Editor-in-Chief September 2015 Ilene Claudius, MD Associate Professor of Emergency Medicine, Keck School of Medicine of USC, Los Angeles, CA Alson S. Inaba, MD, FAAP Associate Professor of Pediatrics, University of Hawaii at Mãnoa John A. Burns School of Medicine, Division Head of Pediatric Ari Cohen, MD Emergency Medicine, Kapiolani Chief of Pediatric Emergency Medicine Medical Center for Women and Services, Massachusetts General Children, Honolulu, HI Hospital; Instructor in Pediatrics, Harvard Medical School, Boston, MA Madeline Matar Joseph, MD, FAAP, FACEP Marianne Gausche-Hill, MD, FACEP, Professor of Emergency Medicine and FAAP Pediatrics, Chief and Medical Director, Professor of Clinical Medicine, Pediatric Emergency Medicine David Geffen School of Medicine Division, University of Florida Medical at UCLA; Vice Chair and Chief, School-Jacksonville, Jacksonville, FL Division of Pediatric Emergency Medicine, Harbor-UCLA Medical Center, Los Angeles, CA Michael J. Gerardi, MD, FAAP, FACEP, President Associate Professor of Emergency Medicine, Icahn School of Medicine at Mount Sinai; Director, Pediatric Emergency Medicine, Goryeb Children's Hospital, Morristown Medical Center, Morristown, NJ Sandip Godambe, MD, PhD Vice President, Quality & Patient Safety, Professor of Pediatrics and Emergency Medicine, Attending Physician, Children's Hospital of the King's Daughters Health System, Norfolk, VA Stephanie Kennebeck, MD Associate Professor, University of Cincinnati Department of Pediatrics, Cincinnati, OH Anupam Kharbanda, MD, MS Research Director, Associate Fellowship Director, Department of Pediatric Emergency Medicine, Children's Hospitals and Clinics of Minnesota, Minneapolis, MN Tommy Y. Kim, MD, FAAP, FACEP Associate Director of Emergency Medicine and Pediatrics, Loma Linda University Medical Center and Children’s Hospital, Loma Linda, CA Melissa Langhan, MD, MHS Associate Professor of Pediatrics, Ran D. Goldman, MD Fellowship Director, Director of Professor, Department of Pediatrics, Education, Pediatric Emergency University of British Columbia; Medicine, Yale School of Medicine, Co-Lead, Division of Translational New Haven, CT Therapeutics; Research Director, Robert Luten, MD Pediatric Emergency Medicine, BC Professor, Pediatrics and Children's Hospital, Vancouver, BC, Emergency Medicine, University of Canada Florida, Jacksonville, FL P. David Sadowitz, MD Associate Professor of Pediatrics and Emergency Medicine, SUNY Upstate Medical University, Department of Emergency Medicine and Pediatrics, Upstate University Hospital, Syracuse, NY Eric Werner, MD, MMM Department of Hematology and Oncology, Children’s Cancer and Blood Disorders Center, Children’s Hospital of The King’s Daughters, Norfolk, VA CME Objectives Upon completion of this article, you should be able to: 1. Describe the deficiencies that result in hemophilia A, hemophilia B, and von Willebrand disease. 2. Identify the bleeding manifestations of hemophilia and apply the appropriate diagnostic workup. 3. Demonstrate appropriate emergency management of hemophilia and von Willebrand disease. 4. Assess how the presence of clotting factor inhibitors modifies management for patients with hemophilia. Prior to beginning this activity, see “Physician CME Information” on the back page. Garth Meckler, MD, MSHS AAP Sponsor Associate Professor of Pediatrics, Martin I. Herman, MD, FAAP, FACEP University of British Columbia; Division Head, Pediatric Emergency Professor of Pediatrics, Attending Physician, Emergency Medicine Medicine, BC Children's Hospital, Department, Sacred Heart Vancouver, BC, Canada Children’s Hospital, Pensacola, FL Joshua Nagler, MD International Editor Assistant Professor of Pediatrics, Harvard Medical School; Fellowship Lara Zibners, MD, FAAP Director, Division of Emergency Honorary Consultant, Paediatric Medicine, Boston Children’s Emergency Medicine, St Mary's Hospital, Boston, MA Hospital, Imperial College Trust; James Naprawa, MD Associate Clinical Professor of Pediatrics, The Ohio State University College of Medicine; Attending Physician, Emergency Department, Nationwide Children’s Hospital, Columbus, OH EM representative, Steering Group ATLS®-UK, Royal College of Surgeons, London, England Pharmacology Editor James Damilini, PharmD, MS, BCPS Clinical Pharmacy Specialist, Emergency Medicine, St. Joseph's Hospital and Medical Center, Phoenix, AZ Joshua Rocker, MD Assistant Professor of Emergency Medicine and Pediatric, Hofstra North Shore-LIJ School of Medicine, Quality Editor Hempstead, NY; Associate Director, Steven Choi, MD Division of Pediatric Emergency Medicine, Cohen Children's Medical Medical Director of Quality, Director of Pediatric Cardiac Inpatient Center, New Hyde Park, NY Services, The Children’s Hospital at Steven Rogers, MD Montefiore; Associate Vice President, Assistant Professor, University of Montefiore Network Performance Connecticut School of Medicine, Improvement; Assistant Professor of Attending Emergency Medicine Pediatrics, Albert Einstein College of Physician, Connecticut Children's Medicine, Bronx, NY Medical Center, Hartford, CT CME Editor Christopher Strother, MD Assistant Professor, Emergency Deborah R. Liu, MD Medicine, Pediatrics, and Medical Assistant Professor of Pediatrics, Education; Director, Undergraduate Keck School of Medicine of USC; and Emergency Department Division of Emergency Medicine, Simulation; Icahn School of Medicine Children's Hospital Los Angeles, at Mount Sinai, New York, NY Los Angeles, CA Case Presentations tion factors VIII and IX, respectively. The overall incidence of hemophilia is 1 per 5000 male births. Hemophilia A is significantly more common than hemophilia B, representing 80% of all hemophilia cases.2 Significant and often life-threatening bleeding (either spontaneous or trauma-induced) is the hallmark of this disease.3 Historically, hemophilia was associated with a dismal prognosis, with most patients dying at a young age of uncontrolled hemorrhage.3 However, life expectancy has increased dramatically in the past 50 years with the advent of plasma-derived and recombinant clotting factor concentrates for treatment of hemophilia. Life expectancy for hemophilia patients in developed countries with comprehensive longitudinal care now approaches that of the general male population.4,5 Von Willebrand disease is significantly more common than hemophilia; however, bleeding in vWD is generally much less severe than that seen in hemophilia. Epistaxis, menorrhagia, oral bleeding, easy bruising, and greater-than-expected postsurgical bleeding are typical presentations of vWD compared to hemophilia, in which more-severe joint, intracranial, and intramuscular hemorrhages are observed.6 Unlike hemophilia, vWD affects men and women equally, with a prevalence as high as 1% of the population, although significantly fewer people will be symptomatic from the disease.7 Von Willebrand disease is not an X-linked disorder, and multiple genetic inheritance patterns exist. Among these, autosomal dominant is the most common pattern. Von Willebrand disease is caused by quantitative or qualitative deficiencies in von Willebrand factor (vWF), a glycoprotein important for platelet aggregation and adhesion to sites of vessel injury.8 Together, vWD and hemophilia A and B represent the vast majority of clinically significant inherited bleeding disorders. Other inherited bleeding disorders do exist and include functional or quantitative deficiencies in clotting factors II, V, VII, X, XI, and XIII, as well as fibrinogen deficiencies. However, these deficiencies, collectively known as the rare bleeding disorders, represent only 3% of all bleeding disorders and each occur with a prevalence of 1 in 500,000.9 Therefore, they will not be addressed in detail within this review. Acquired bleeding disorders also will not be reviewed here. In patients diagnosed with hemophilia, ED visits are frequent. Not surprisingly, the majority of these visits are for acute bleeding, where prompt and appropriate intervention by an emergency clinician is critical in mitigating morbidity and mortality.10,11 In some cases, a new diagnosis of a bleeding disorder may be first identified in the ED.11,12 However, several lines of evidence indicate that many emergency clinicians have little experience and lack familiarity in evaluating and managing patients with the common inherited bleeding disorders.1,10 Expert guidelines for the evaluation and management of patients with inherited bleeding disorders exist,13-15 but such A 7-year-old boy with a history of severe hemophilia B, who receives scheduled prophylactic factor IX 3 times a week, presents to the emergency department after rolling off a bed 3 feet from the ground and striking his head. He complains of a mild headache at this time. On examination, there is no palpable scalp hematoma, and the neurologic examination is entirely normal. The patient’s last prophylactic factor infusion was 2 days ago. Given his normal examination at this time, is head imaging indicated? Should he receive a factor IX infusion? If so, when should it be administered, and how much should be given? The patient uses AlphaNine® SD brand factor IX for his factor replacement at home, but your emergency department only stocks BeneFIX® brand factor IX. Is it okay to switch factor products? Are there risks to doing so? A 17-year-old adolescent boy with a history of severe hemophilia A with a high-titer inhibitor presents to the emergency department with pain and swelling in his right knee that developed at school that day. There is no history of antecedent trauma. Examination of the affected knee reveals a large effusion and pain with flexion > 90°. Are there laboratory studies or imaging that could aid in diagnostic decision-making? Should you perform arthrocentesis either diagnostically or therapeutically? Should you treat with a factor VIII concentrate? How does the presence of a high-titer inhibitor change your management? Your ED stocks both FEIBA and recombinant factor VIIa as bypassing products. Is one better than the other for this patient? Can this patient be discharged from the emergency department after treatment or does the presence of inhibitors preclude home management? A 16-year-old girl presents to the emergency department with a 1-month history of menorrhagia. Her primary care physician has seen her for this complaint previously and has sent laboratory tests, including a complete blood count and prothrombin time/partial thromboplastin time, which were normal. Her von Willebrand panel was significant for a low von Willebrand factor antigen level (vWF:Ag) of 20 IU/dL and a low ristocetin cofactor activity level (vWF:RCo) of 22 IU/dL. She has not started any treatment. What is her likely diagnosis, and what treatment options are available for this patient to decrease menorrhagia? Should she receive oral contraceptive pills, desmopressin, and/or aminocaproic acid? What risks are associated with these therapies? Should her family members seek testing for bleeding disorders? Introduction Hemophilia and von Willebrand disease (vWD) represent the most common inherited bleeding disorders, and, consequently, those most likely to be encountered in the emergency department (ED).1 Hemophilia A and B are X-linked recessive disorders caused by the deficiency or absence of coagulaCopyright © 2015 EB Medicine. All rights reserved. 2 Reprints: www.ebmedicine.net/pempissues Defining vWD has been challenging for the purpose of creating cohorts of subjects.7 Many patients with abnormal testing on von Willebrand laboratory panels have no clinical manifestations of bleeding, and, yet, some patients with equivocal testing on von Willebrand panels are significantly symptomatic.8 These complications have made accurate identification of a study population more challenging. Because of the rational basis for giving factor concentrates in cases of acute bleeding in pediatric patients, the relatively low risk of doing so in the era of recombinant factor concentrates, and excellent infection screening for plasma-derived products, a randomized trial withholding factor concentrates for patients with acute bleeds is not feasible, nor is it appropriate. recommended standards are often not met in the ED care of patients with hemophilia.16 Thus, evidencebased knowledge of ED evaluation and management of inherited bleeding disorders is critical. Critical Appraisal Of The Literature A literature search was performed in PubMed using the search terms hemophilia, factor VIII deficiency, factor IX deficiency, Christmas disease, children, and pediatric. This search yielded 5999 results that were evaluated by the authors, with 118 papers deemed relevant to this review of hemophilia A and B. A separate search was conducted using the search terms von Willebrand disease, children, and pediatric. This search yielded 1132 results which were reviewed, identifying 30 relevant references for vWD. The Cochrane Database of Systematic Reviews was searched using the key terms hemophilia and von Willebrand disease. This search yielded 12 results, 3 of which were relevant to this ED-focused review. A search of The National Guideline Clearinghouse (www.guideline.gov) yielded 2 sets of guidelines for hemophilia from the World Federation of Hemophilia and the British Committee for Standards in Haematology, and 1 guideline for vWD from the National Heart, Lung, and Blood Institute of the National Institutes of Health. Most recommendations regarding acute treatment of inherited bleeding disorders contained within the hemophilia guidelines are level IV (based on case series/historically controlled studies), with the exception of recommendations for factor prophylaxis, which have stronger evidence (level II, based on randomized controlled trials). For vWD, recommendations are largely derived from case series and retrospective reports (almost all grade C recommendations with level IV evidence). The absence of a significant number of large randomized trials for hemophilia is multifactorial. Modern hemophilia care began with the development of plasma-derived factor concentrates in the 1960s. Subsequent research was confounded by the staggering number of patients with hemophilia who were affected by HIV infections contracted via plasma-derived clotting factor infusions in the late 1970s and early 1980s.17 A 1998 study by Rosenberg and Goedert estimated that nearly half of all patients with hemophilia living in the United States were infected with HIV.18 Despite being the most common inherited clotting factor deficiency, hemophilia remains a rare disease, and randomized controlled trials generally require multicenter participation to accrue an adequate number of patients. Modern national registries for patients with hemophilia have aided in consolidating retrospective information on these patients, but they have not effectively facilitated a large number of prospective multicenter trials.19 September 2015 • www.ebmedicine.net Etiology And Pathophysiology Hemostasis And The Coagulation Cascade Before discussing the specific pathophysiology underlying vWD and hemophilia, an overview of the normal bleeding cessation and clot formation process is useful. Hemostasis is generally divided into primary and secondary responses. Primary hemostasis involves the formation of a platelet plug, whereas secondary hemostasis involves the coagulation cascade and fibrin reinforcement of the primary platelet plug.20 The primary hemostatic response begins with local vasospasm to decrease hemorrhage. Next, circulating platelets adhere to newly exposed vascular collagen and to each other, a process that is mediated by vWF. This is followed by platelet activation and platelet aggregation. The end result of primary hemostasis is a platelet plug over the site of vascular injury. Unfortunately, this plug is short-lived and is easily sheared when normal blood flow returns.20,21 Therefore, secondary hemostasis is required. This step utilizes the coagulation cascade, which includes a variety of proenzymes and their cofactors interacting on damaged endothelium or platelets. The cascade occurs in 3 phases: initiation, amplification, and propagation. Initiation occurs with the release of tissue factor (TF) from damaged endothelium. TF binds to factor VII, and this complex, in turn, activates factor X and factor IX. Factor X combines with factor V, which converts prothrombin to thrombin (factor II). However, this process only activates a small amount of thrombin. In order to produce more thrombin, a positive feedback loop known as the amplification phase is initiated by thrombin itself. In this phase, thrombin activates the production of more factor V, factor VIII, factor IX binds with factor VIII, which, in turn, activates the production of even more factor X-factor V complex. This leads to an even greater amount of thrombin. In addition to the roles just described, thrombin converts fibrinogen to fibrin. Factor XIII covalently crosslinks the fibrin 3 Mobile app access: www.ebmedicine.net/app strands, strengthening the thrombus and completing the coagulation cascade.20 cases per 1 million patients. It is caused by a severe quantitative defect in vWF, with undetectable levels of vWF leading to markedly decreased levels of factor VIII. These patients have bleeding phenotypes similar to hemophilia A.6 In addition to the various subtypes of vWD, there exists a subset of patients with vWF:Ag or vWF:RCo in an intermediate range (30-60 IU/dL) who will have clinical bleeding symptoms. These patients are sometimes referred to as “low vWF,” and they are potentially eligible for the same treatments administered for vWD. Pathophysiology Of Von Willebrand Disease Von Willebrand disease is the most common bleeding disorder in the United States. However, its clinical manifestations vary greatly, and it is thought that only 10% of patients with laboratory-defined vWD will have symptomatic disease.22 Von Willebrand disease is defined by a qualitative or quantitative defect in vWF. This glycoprotein is synthesized in endothelial cells and megakaryocytes, and it is secreted into circulation, where it participates in hemostasis in 3 ways: (1) vWF binds to platelet glycoprotein receptor GPIb/IX, which initiates platelet adhesion to injured subendothelium; (2) vWF interacts with the GPIIb/IIIa receptor on platelets, which facilitates platelet aggregation; and (3) vWF binds and stabilizes factor VIII in circulation, preventing it from being degraded by protein C.8,21 Three categories of vWD exist, and they vary in pathophysiology and phenotype. Type 1 vWD accounts for 80% of patients with vWD and is defined by a quantitative defect in vWF.21 There is much debate about the precise cut point for diagnosis, but expert opinion suggests a vWF antigen (vWF:Ag) level, a quantitative measure of vWF, of < 30% to 40% of normal, along with a history of bleeding symptoms. Such patients will also generally have decreased performance on the vWF ristocetin cofactor (vWF:RCo) activity assay (< 30), a measure of vWF function. Type 2 vWD is due to a qualitative defect of vWF, of which there are 4 subtypes. Type 2A occurs in 10% of patients with vWD, and it is caused by a mutation in vWF and the subsequent inability of vWF to form high-molecular-weight multimers. These multimers are necessary for the optimal functionality of vWF. Patients with type 2A vWD have decreased performance on vWF:RCo assay, but may have a normal quantity of vWF as measured by vWF:Ag. Type 2B disease is caused by a gain of function mutation in the GPIb binding site on vWF, such that it binds with too much avidity to platelets. This decreases highmolecular-weight multimers and leads to platelet agglutination, and, occasionally, thrombocytopenia, as platelets precipitate from the blood. A decreased vWF:RCo is observed in these patients. Type 2M vWD is a family of vWF abnormalities that cause structural defects in the molecule that decrease its multimeric size. These defects may lead to altered vWF binding to platelets or to adhesive proteins such as type 3 collagen. Type 2N vWD is caused by the inability of vWF to bind to factor VIII. Therefore, factor VIII is easily degraded in circulation, and patients will have decreased factor VIII levels. As a consequence, they typically present with symptoms similar to a patient with hemophilia A. The third subtype of vWD is type 3. This subtype is quite rare, with only 1 to 2 Copyright © 2015 EB Medicine. All rights reserved. Pathophysiology Of Hemophilia Hemophilia is an X-linked recessive disorder, and it is divided into hemophilia A and B. Hemophilia A is defined by a deficiency in factor VIII, and it occurs in 1 in 5000 males. Hemophilia B is due to a deficiency in factor IX, and it occurs in 1 in 30,000 males.20 Hemophilia has no racial or geographic predilection.23 One-third of hemophilia cases are caused by de novo mutations, and, therefore, a patient with this diagnosis may not have a family history of the disease.24 The absence of factor VIII or IX leads to the inability to activate factor X and the subsequent inability to activate thrombin. Therefore, both hemophilia A and B have similar phenotypes and, on laboratory testing, present with prolonged partial thromboplastin time (PTT). Both diseases are further characterized by their severity, which is defined by the level of clotting factor activity. Patients with severe disease have < 1% of normal clotting factor activity, patients with moderate disease have 1% to 5% of normal clotting factor activity, and patients with mild disease have 5% to 40% of normal clotting factor activity. Patients with mildly decreased levels of factor VIII or IX with > 40% of normal clotting factor activity are unlikely to be symptomatic, and they are not generally considered to have a clinical diagnosis of hemophilia.23 In hemophilia A, 35% of patients have severe disease, 15% have moderate disease, and 50% have mild disease. In contrast, 60% of patients with hemophilia B have severe disease.14 The severity of disease helps predict the bleeding phenotype. Patients with severe disease often have multiple bleeding episodes per month, usually in joints and soft tissues, and they can also have spontaneous bleeds. Patients with moderate disease typically bleed 4 to 6 times per year and will bleed with mild trauma. Mild disease does not generally cause frequent bleeding episodes, and these patients may only hemorrhage with surgery or significant trauma, though correlation of bleeding symptoms with baseline factor levels is not consistent, and some patients with baseline factor levels > 1% will nonetheless have severe bleeding.25 4 Reprints: www.ebmedicine.net/pempissues Differential Diagnosis mophilia and vWD should invoke consideration of these diagnoses in the undiagnosed patient. The most common screening tests for a patient with a suspected bleeding disorder include a complete blood count (CBC), prothrombin time (PT), and PTT. Typically, hemophilia A and B both cause isolated prolongation of the PTT with a normal PT and platelet count. When a prolonged PTT is noted in a male patient with significant bleeding, assays for levels of factor VIII and IX should be ordered. The differential diagnosis for isolated prolongation of PTT includes laboratory or sampling errors (such as inadequate specimen volume in a sample tube or a partially clotted sample), the presence of heparin in the patient, in the line from which the sample was drawn, or in the tube in which the sample was sent, and factor XI deficiency (sometimes called hemophilia C, which is extremely rare). In addition, factor XII deficiency and the presence of the lupus anticoagulant will result in a prolonged PTT on laboratory testing, but do not result in a clinical bleeding phenotype. An isolated PT prolongation is caused by factor VII deficiency. Prolongation of both PT and PTT results from factor V, factor X, fibrinogen, or prothrombin deficiencies.25 Depending on the severity and the degree to which factor VIII levels are affected, vWD may or may not result in prolonged PTT. Platelet count will generally be normal, though studies of platelet function (eg, PFA-100 or platelet aggregation studies) will be abnormal.21 Table 1 presents the differential diagnosis for bleeding disorders based on abnormalities in PT and PTT studies. Hemarthrosis Hemarthrosis is the classic manifestation of hemophilia and the most common bleeding symptom in patients with hemophilia. It can occur up to 20 to 30 times per year in patients with severe hemophilia.28 Patients generally report mild pain in the affected joint that progresses to severe pain, swelling, warmth, and decreased range of motion, with the ankles, knees, and elbows being most commonly affected.3 While hemarthrosis is not the most common initial presenting symptom of hemophilia, spontaneous joint bleeding should trigger workup for a bleeding disorder.26,29 Hematomas/Soft-Tissue Bleeds Hematomas are a common initial presentation in hemophilia. Cohort studies suggest that 17% to 40% of infants with hemophilia, particularly mild to moderate hemophilia, may present with large soft-tissue hematomas.30 Features that may suggest a bleeding disorder include hematoma formation after minimal trauma and slow resolution of the lesions.26,31 Muscle hematomas are the second most common bleeding manifestation in hemophilia, behind only hemarthroses, and they are a more common initial presentation of the disease. These bleeds generally present with muscle swelling, decreased range of motion, pain, a decrease in hemoglobin, and possible nerve entrapment.32 Mucosal Bleeding Both patients with vWD and patients with hemophilia can present with prolonged or severe mucosal bleeding. Adolescent females with vWD classically Timing Of Diagnosis While children with hemophilia and vWD can have classical clinical presentations, it is important to recognize that young children and children with milder disease may not carry a diagnosis at the time of presentation to the ED. In a German cohort of children with severe hemophilia A, only 44% had a bleeding episode during the first year of life. A few children did not have a bleed until after age 4 years, with an average age of diagnosis of 1.2 years.26 A retrospective cohort study of 55 patients calculated an average age of diagnosis of 5.3 years in children with mild hemophilia and no known family history of disease.27 A British survey of the caregivers of 12 children with hemophilia suggested that patients often seek medical attention multiple times (either in the ED or from their primary care physician) before receiving a diagnosis.12 Accurate diagnosis of the underlying bleeding disorder is important both in acute management and in the prevention of longterm morbidity and mortality. Table 1. Differential Diagnosis For Bleeding Disorders By Abnormality In Prothrombin Time/Partial Thromboplastin Time Studies Typical Presenting Symptoms Of Inherited Bleeding Disorders At Diagnosis The following typical bleeding presentations of heSeptember 2015 • www.ebmedicine.net Laboratory Finding Differential Diagnosis Prolonged PT only • Factor VII deficiency • Liver failure Prolonged PTT only • • • • Prolonged PT and PTT • • • • • Factor VIII, IX, XI, or XII deficiency Lupus anticoagulant Heparin in sample von Willebrand disease (with low factor VIII) • Specimen integrity problems Factor V or X deficiency Fibrinogen or prothrombin deficiency Disseminated intravascular coagulation Lupus anticoagulant Specimen integrity problems Abbreviations: PT, prothrombin time; PTT, partial thromboplastin time. 5 Mobile app access: www.ebmedicine.net/app present with menorrhagia (defined as bleeding for > 7 days or loss of > 80 mL of blood per menstrual cycle).9 Younger children with vWD can have excessive or prolonged bleeding from mucous membranes (such as occurs in epistaxis or in bleeding after dental procedures). Children with hemophilia may also present with similar symptoms, but they can also have more-severe mucosal bleeding, such as gastrointestinal hemorrhage.21 crutches for weight-bearing joints) to help prevent further damage.16 If blood loss is significant, volume should be repleted with crystalloid. Most hematologists recommend initiating factor replacement within 1 hour of bleeding. Therefore, if home factor infusions cannot be completed, the patient should be taken to a treatment center expeditiously.38 Intracranial Hemorrhage Intracranial hemorrhage (ICH) is the most serious complication in children with bleeding disorders. ICH has been reported to have 20% mortality in the hemophilia population, though this rate is lower in patients aged < 19 years. Among surviving patients, 25% to 75% sustain permanent disability.33 ICH is 20 to 50 times more frequent in males with hemophilia than in males without. ICH is a less-frequent complication in patients with vWD, but no large studies have been conducted on this subject. Case series suggest that patients with the rare type 3 subtype of vWD are at higher risk for ICH.34 While ICH is certainly seen in patients without inherited bleeding disorders, ICH with minimal or no antecedent trauma raises the suspicion of an underlying bleeding disorder, and screening laboratory testing should be considered in these patients.35 The child with a bleeding disorder requires a unique approach in the ED, and enlisting the support of a hematologist is very important. However, not all patients with a primary coagulation defect come to the ED with a known diagnosis. As stated previously, one-third of all patients with hemophilia have a de novo mutation, and have no known family history. In fact, a British study of 25 patients with hemophilia without a known family history suggested that the mean age of diagnosis is 29.5 months.12 Patients with milder bleeding disorders tend to be diagnosed later in life. In older children, menorrhagia may be the first or only sign of vWD.1 Therefore, it is important to have a high index of suspicion in children with multiple episodes of easy bruising or bleeding. The initial evaluation is driven by vital signs and pertinent presenting symptoms and examination findings. The appropriate resuscitative measures take precedence over disease-specific measures. Perhaps the only variation on the secondary survey is immobilization of any bleeding joint to prevent further injury.16 It is only once the child is stabilized that bleeding disorder evaluation and management should be prioritized. There is no universally agreed-upon algorithm for evaluating a patient with a bleeding disorder. However, there are guidelines from the United Kingdom Haemophilia Centre Doctors' Organisation that can help guide the initial assessment.16 (See Table 2, page 7.) After a complete history is taken (including the elements noted in Table 2) and a complete physical examination is completed, diagnostic studies can be considered. Emergency Department Evaluation Postsurgical Bleeding Prolonged or excessive bleeding after minor surgical procedures is a cardinal feature of both hemophilia and vWD, and it is often the presenting symptom at diagnosis.36 Many boys with hemophilia are diagnosed when they have significant bleeding after circumcision. It is important to realize, however, that this will not always occur in patients with hemophilia and will occur only very rarely in patients with vWD. In a cohort of boys with severe hemophilia A, 50% underwent circumcision without any bleeding complications.26 Prehospital Care Diagnostic Studies Optimizing home treatment in patients with bleeding disorders has been shown to decrease total factor concentrate utilized and subsequent cost to the healthcare system.37 Unfortunately, there is little research on prehospital care in this population, and current recommendations are based primarily on expert opinion.38 Most patients with severe hemophilia receive prophylactic factor infusions in the home to prevent bleeds. Additional doses are administered in response to bleeding symptoms. Other prehospital care might include ice, compression, and elevation for joint or soft-tissue bleeds.39 In cases of hemarthrosis, the joint should be protected (eg, using Copyright © 2015 EB Medicine. All rights reserved. Most recommendations for imaging and diagnostic studies in a child with a bleeding disorder are based upon expert consensus, given the relative dearth of data. In the ED, the workup is primarily dependent upon the presenting complaint and the patient’s mental status and vital signs. Laboratory Studies Order a blood type and crossmatch and a CBC to assess hemoglobin and platelets in children with severe bleeds and/or alterations in vital signs. Coagulation studies (PT, PTT, and fibrinogen) can 6 Reprints: www.ebmedicine.net/pempissues be useful if the diagnosis of a bleeding disorder is being considered. However, these studies will not detect patients with qualitative platelet disorders, many forms of vWD, and the rare factor XIII deficiency. For patients with low platelet levels and normal PT/PTT levels, consider and work up systemic causes of thrombocytopenia, such as idiopathic thrombocytopenic purpura, hemolytic uremic syndrome, autoimmune disorder, bone marrow disease, etc. In patients with hemophilia, baseline factor levels (factor VIII in hemophilia A, factor IX in hemophilia B) can help emergency clinicians determine goal treatment levels; however, the results of these tests will generally not be available while the patient is in the ED, and cannot be used to guide treatment. Therefore, factor dosing must be chosen empirically. If the patient has a known diagnosis of factor VIII or factor IX deficiency but the severity is unknown, it is reasonable to assume that the patient has severe disease for the purpose of factor dosing. If a baseline level has not been established and/or the patient has received treatment for the disorder, order a factor VIII level in patients with vWD and symptoms of severe bleeding.40 Inhibitor titers in the ED setting are not useful in acute management, as results are generally not available while the patient is in the ED, but they may help guide ongoing therapy if the patient is suspected of having new development of inhibitors. Factor levels can be measured 30 to 60 minutes after factor infusion to determine the patient’s response to the infusion. If there is little increase in the measured factor level, an inhibitor is likely present. The utility of vWD screening in the ED is limited, as vWF is an acute phase reactant and any measurements of vWF antigen during acute illness can be potentially misleading. In patients in whom vWD is suspected based on clinical or family history, consider a vWF:Ag level, vWF:RCo activity level, and a factor VIII activity level. However, these should be interpreted with caution as false negatives are common in the setting of acute bleeding or illness and estrogen or corticosteroid use. Patients with negative tests in whom the index of suspicion remains high should have repeat testing in the outpatient setting. Imaging Studies Regardless of the need for imaging, the process of obtaining imaging should not delay factor replacement. Head Imaging There are no consensus guidelines as to which patients with head trauma require imaging and which patients can be safely observed.41 Two retrospective cohort studies have been completed looking at the use of head computed tomography (CT) scans in patients with hemophilia with concern for ICH. One study at the Children’s Hospital of Philadelphia found 9 cases of ICH. In 5 of these 9 cases, the patients were asymptomatic at presentation, leading the authors to suggest that early, more liberal use of head CT scans may be justified.42 However, another retrospective cohort study at The Hospital for Sick Children in Toronto identified 11 cases of ICH in children with hemophilia. In all 11 cases, the patients presented with altered mental status or signs of increased intracranial pressure.43 Given this conflicting evidence, it seems reasonable to obtain a head CT or magnetic resonance imaging (MRI) scan in patients with severe hemophilia after any head trauma, particularly in infants, patients with severe disease, or patients with inhibitors. All patients with altered mental status or an abnormal neurological examination require emergent head imaging. If possible, an MRI is preferred, given its ability to better visualize the posterior fossa.43 In considering ICH in vWD, a retrospective cohort study was performed in children with vWD and head trauma. The study identified 6 children with ICH, all of whom presented with signs of elevated intracranial pressure and/or altered consciousness. Therefore, it seems reasonable to reserve Table 2. Summary Of The United Kingdom Haemophilia Centre Doctor’s Organisation Evaluation Guidelines16 1. The patient should be assessed within 15 minutes of arrival. Treatment, if indicated, should be initiated within 30 minutes of arrival. 2. Emergency clinicians should know or have immediate chart access to the patient’s diagnosis and the severity of the bleeding disorder. The clinician should also know if the patient has inhibitors. 3. It is useful to know what factor product the patient generally responds to and/or uses at home. Many hemophilia treatment centers provide their patients with a summary letter that contains the specific treatment product(s) and dosage recommendations. 4. For patients with vWD or mild hemophilia A, a history of responsiveness or nonresponsiveness to desmopressin should be obtained. 5. The emergency clinician should determine whether the patient has had any adverse events due to the bleeding disorder and any adverse reactions to factor concentrates. In the older patient, it is useful to know whether the patient has contracted any bloodborne diseases (such as hepatitis C or HIV), as this may broaden the consideration of potential problems and comorbidities associated with more-severe and more-frequent bleeding sequelae. 6. The emergency clinician should contact the primary hematologist for any bleeding episode. The primary hematologist will be able to help guide management decisions and establish follow-up, though treatment can be initiated prior to discussion with hematology. 7. If a significant bleed is suspected, the patient should be stabilized and, if possible, transferred to a hemophilia treatment center, as treatment at these centers is associated with improved outcomes. Treatment for the bleed should be initiated as soon as possible. Abbreviations: HIV, human immunodeficiency virus; vWD, von Willebrand disease. September 2015 • www.ebmedicine.net 7 Mobile app access: www.ebmedicine.net/app imaging in vWD only to symptomatic patients or patients with a particularly concerning mechanism of injury. Exceptions include patients with severe vWD (type 2N or type 3) whose phenotype is more similar to hemophilia and who should undergo imaging more liberally.34 factor IX in the recipient’s plasma by 1% (0.01 IU/mL).3 However, the patient’s individual response may vary, so peak levels should be confirmed for major or lifethreatening bleeds. Different types of bleeding require different dosing and duration of factor infusions to achieve adequate hemostasis. Factor concentrate vials vary in the exact number of units of clotting factor contained in each vial. The number of units of factor is labeled on each vial, and dosing should be rounded to the nearest number of vials, rounding up when necessary. Except in young infants, where the available vials of factor concentrates may be limited, it is unlikely that a partial vial dose would be indicated. Table 4 (see page 9) provides a quick reference for dosing and disposition of hemophilia patients by specific bleeding sites. Joint And Soft-Tissue Imaging Hemarthrosis is usually clinically apparent. Therefore, no diagnostic imaging is necessary if a patient with a known bleeding disorder presents with joint swelling and pain, particularly in a known target joint.44 However, ultrasound can be used if the diagnosis is in doubt, and this modality is quite sensitive for detection of hemarthrosis.45 Plain x-ray films may be indicated if there is a significant traumatic mechanism and concern for fracture. Soft-tissue bleeding may require diagnostic imaging in the ED. Very superficial hematomas are clinically apparent, and imaging is unnecessary in these cases unless there is concern for underlying bony injury. Deeptissue injuries (such as bleeds into muscles) will often require imaging for definitive diagnosis and baseline measurements. Ultrasound is useful for most soft-tissue hematomas, and CT or MRI can be used if a deeper softtissue bleed is suspected.30,46 Management Of Specific Types Of Bleeding In Hemophilia Hemarthrosis Up to 85% of bleeds in patients with hemophilia occur in joints, and ankles, knees, and elbows are most commonly affected.3 If a single joint becomes a site of recurrent hemarthrosis, it is termed a target joint. Over time, synovitis can occur in such joints and, if uncontrolled, this can lead to cartilage destruction and significant functional impairment from hemophilic arthropathy.55 Therefore, significant effort should be directed toward minimizing bleeding into joints with the goal of preservation of long-term joint function as well as averting short-term pain and disability. In the setting of acute hemarthrosis, consensus guidelines recommend early factor infusion with an anticipated goal factor VIII or IX level of 40% to 60%, based on predicted response to factor infusion as well as symptom improvement, although postinfusion Treatment Treatment Of Bleeding From Hemophilia Treatment for hemophilia is directed toward replacement of the missing clotting factor (factor VIII replacement for patients with hemophilia A and factor IX replacement for patients with hemophilia B). There are 2 general types of clotting factor concentrates available: (1) Plasma-derived factor concentrates, made up of pooled human donor plasma; these have been available since the 1960s; and (2) recombinant factor concentrates, which are manufactured in mammalian cell culture systems, and have been available since the 1980s.17 Both recombinant and plasma-derived factor concentrates have been shown to be highly effective in achieving hemostasis, with a success rate of ≥ 90% after 1 to 2 doses of factor concentrates for typical bleeding episodes.47,48 There are no quality studies proving that switching from one type of factor product to another or among different brands of factor products increases the risk of inhibitor development; however, this should generally be avoided, if possible.49,50 Therefore, in situations where a patient’s specific brand of factor is unavailable, and urgent treatment is necessary, another brand of factor can be utilized.51 Table 3 details the currently available factor replacement products. In general, 1 unit/kg of factor VIII will increase factor VIII in the recipient’s plasma by 2% (0.02 IU/ mL), whereas 1 unit/kg of factor IX will increase the Copyright © 2015 EB Medicine. All rights reserved. Table 3. Available Factor Replacement Products52 Factor VIII Concentrates Recombinant • Advate, Helixate® FS, Kogenate® FS, Recombinate, Xyntha®, Novoeight®, Eloctate® Plasma-Derived • HEMOFIL M, MONARC-MTM, Monoclate-P® Factor IX Concentrates Recombinant • BeneFIX®, Alprolix® Plasma-Derived • AlphaNine® SD, Mononine® von Willebrand Factor Concentrates (with FVIII) Plasma-Derived • Alphanate®, Humate-P®, Wilate® Bypassing Agents • FEIBA NF, NovoSeven® RT 8 Reprints: www.ebmedicine.net/pempissues Muscle Bleeding levels will not generally be measured. In many cases of hemarthrosis, a single dose of factor replacement is adequate; however, if symptoms have not improved after 1 dose, repeat doses may be added at 12-hour to 24-hour intervals until the joint normalizes.44 In addition, immobilization of the affected joint, use of a compression bandage, and application of ice is recommended, though significant evidence for these recommendations is lacking.14 Ice, if used, should be applied for 15 minutes every 2 to 3 hours. Analgesia is also important. Nonsteroidal anti-inflammatory drugs should be avoided in patients with hemophilia due to concern for adding antiplatelet effects in a patient with existing coagulopathy. In general, acetaminophen, with or without an opioid, is used for pain control, though guidelines recommending this are not well researched.54 Corticosteroids are generally not recommended for pain control, and there is no evidence that they provide significant benefit in control of pain or prevention of arthropathy.56 Guidelines do not recommend aspiration or arthrocentesis for management of hemarthrosis, and there is very little literature addressing this subject. The second most common type of bleeding seen in patients with hemophilia is bleeding into muscle. Factor should be infused as soon as possible to target predicted levels of 40% to 60%. Repeat infusions are often required every 12 to 24 hours for 2 to 3 days, along with careful monitoring (often as an inpatient), as compartment syndrome or nerve compression can develop, and significant rehabilitation/physical therapy may be required, which can precipitate rebleeding.14,57 Iliopsoas Hemorrhage One particular type of muscle bleed that merits special mention is iliopsoas muscle hemorrhage. In comparison to other muscle hemorrhages, iliopsoas hemorrhage has the potential for massive blood loss and represents a potentially life-threatening condition in patients with hemophilia. Motor limitations can also be severe.58 In the 3 largest published series of iliopsoas hemorrhage (all of which were retrospective chart reviews), the most common presentations involved thigh, hip, abdomen, and/or groin pain. A flexion contracture at the hip was noted in the vast majority of patients in all series, and paresthesia in the femoral nerve distribution was a classic Table 4. Dosing And Duration Of Therapy For Hemophilia-Related Bleeds1,14,44,53,54 Type of Bleeding Goal Factor Level Hemophilia A (IV dose of Factor VIII) Hemophilia B (IV dose of Factor IX) Duration of Treatment Disposition from ED/Special Notes Hemarthrosis 40%-60% 20-40 units/kg 40-60 units/kg 1-3 days (1 dose is usually sufficient) Single dose is often adequate, patient can usually discharge home with splinting, ice, and close follow-up Muscle hemorrhage (except iliopsoas) 40%-60% 20-40 units/kg 40-60 units/kg 2-3 days, repeat doses with physical therapy Admission is occasionally required to monitor for compartment syndrome; physical therapy is recommended Iliopsoas bleed 80% 50 units/kg 80-100 units/kg 7-14 days, maintain levels > 80% for 3 days, then > 50% thereafter Admit to hospital, strict bed rest, monitor serial Hct, follow-up imaging with CT, MRI, or ultrasound Major trauma or surgery 100% 50 units/kg 100 units/kg 14 days, maintain > 80% for days 1-3, then > 40% for days 4-6, and > 30% for days 7-14 Give factor as soon as possible, prior to imaging if any delay is anticipated; trauma surgery consultation, admission Intracranial hemorrhage 100% 50 units/kg 100 units/kg 14 days, maintain > 80% for days 1-7, and > 50% thereafter Give factor before obtaining imaging as soon as ICH is suspected; neurosurgery consultation, ICU admission Hematuria/renal hemorrhage 50% 20-30 units/kg 50-60 units/kg 1-3 days Admission, hydrate aggressively (3 L/m2 x 48 h), avoid antifibrinolytics, consider urology consultation Gastrointestinal bleed 100% 50 units/kg 100 units/kg 7-10 days, maintain > 80% for days 1-6, and > 30% for days 7-10 Admit, consider antifibrinolytics, gastroenterology consultation Deep laceration 50% 25 units/kg 50 units/kg Up to 5-7 days, depending on depth/location Give factor, then suture, discharge home, close follow-up within 24 hours Abbreviations: CT, computed tomography; ED, emergency department; Hct, hematocrit; ICH, intracranial hemorrhage; ICU, intensive care unit; IV, intravenous; MRI, magnetic resonance imaging. September 2015 • www.ebmedicine.net 9 Mobile app access: www.ebmedicine.net/app ICH. This should occur prior to obtaining imaging, if imaging is anticipated to delay factor administration in any way.14 sign seen in many patients.58-60 In one of these series of 14 iliopsoas bleeds, hip flexion contracture was present in 13 of the 14 patients, and femoral paresthesia was present in 8 of the 14 patients.59 Bleeding can occur spontaneously, without trauma, and delay in seeking care is common. In a series of 46 episodes, the duration of symptoms prior to seeking medical attention was 3.8 days, and only 57% of the patients reported a history of trauma.59 The initial factor treatment goal for patients with iliopsoas bleeds is higher than that for other muscle bleeds. The goal is a predicted postreplacement level of 80% for at least 3 days, and then a goal of > 50% for the remaining duration of treatment.59 Treatment duration is typically 7 to 14 days. Expert guidelines recommend that patients should be admitted to the hospital and placed on strict bed rest to avoid any significant motion at the hip, contracting the psoas muscle and potentially exacerbating the condition.14 MRI (preferred), ultrasound, and CT scanning can all be considered to aid in diagnosis, and can be used for follow-up imaging as the patient recovers.14 Intracranial Hemorrhage In Neonates Intracranial hemorrhage in patients with hemophilia represents a significant challenge, and there is debate regarding head imaging in neonates with known hemophilia. Some guidelines suggest screening imaging by ultrasound or CT for all newborns with a family history of hemophilia plus an instrument-assisted delivery.64 However, as 30% of cases of hemophilia have no family history, a high index of suspicion for hemophilia is necessary for infants with unusual intracranial hemorrhages in the neonatal age group. Major Trauma In Hemophilia In settings of major trauma or where major emergency surgery may be indicated, immediate factor replacement to a goal level of 100% is recommended, with a tapering goal factor level over the days following as described in Table 4 (see page 9).14 Imaging should not delay factor administration. Further, in any patient with hemophilia and peritoneal signs, bleeding should be suspected regardless of whether there is a history of preceding trauma.53 Intracranial Hemorrhage ICH is a major source of mortality in patients with hemophilia, with an overall mortality rate of 9% to 39%, though this is lower in the pediatric population.42 It is also a major source of long-term disability and decreased quality of life in patients with hemophilia.61 Furthermore, ICH is quite common, with a prevalence rate of 2% to 12% in pediatric patients with hemophilia.41,62 The 3 most commonly associated risk factors for ICH across multiple studies are: (1) severe hemophilia; (2) the presence of inhibitors; and (3) a history of trauma.33,41 While trauma is a major risk factor for ICH, spontaneous ICH can occur. For example, in a series from Brazil, a history of trauma was reported in just 21 of the 45 patients who presented with intracranial bleeding.35 The only sign consistently correlated with the presence of ICH is a change in mental status. In a series from the Children’s Hospital of Philadelphia including 374 ED visits, only drowsiness and loss of consciousness were identified as statistically significant predictors of ICH.42 However, the absence of mental status changes does not exclude ICH. A neurologic deficit or altered mental status was only present in 38% and 33% of cases, respectively, in the series of patients with hemophilia with ICH from Brazil. Furthermore, 44% of patients in that series showed no abnormalities on physical examination.35 The prompt administration of factor is critical in cases of potential ICH, and factor administration within 6 hours of head trauma has been demonstrated to reduce the risk of ICH and lower mortality rate in patients with hemophilia.63 Factor should be administered immediately in patients with hemophilia who present with head trauma or any signs or symptoms that raise suspicion for spontaneous Copyright © 2015 EB Medicine. All rights reserved. Less Common Bleeds In Patients With Hemophilia Gastrointestinal Hemorrhage Gastrointestinal hemorrhage is a less common site of bleeding in patients with hemophilia, and it may represent comorbid gastrointestinal disease. Initial factor replacement should target factor levels of 80% to 100% for 1 to 6 days, followed by maintenance replacement with a goal factor level of 50% for 7 to 14 days. Gastroenterology consultation is recommended to better identify the source of the hemorrhage.14 Hematuria/Renal Hemorrhage For patients presenting with gross hematuria and evidence of upper renal tract hemorrhage, vigorous hydration at 1.5 times the maintenance fluid requirement for 48 hours is recommended.14 Factor replacement to a goal of 50% for 3 to 5 days should be provided, and urology consultation should be considered. Antifibrinolytic agents (eg, aminocaproic acid, tranexamic acid) are contraindicated in these patients as they can precipitate thrombi within the kidneys.14 Prophylaxis In Hemophilia There is a significant body of literature to support the use of regular, scheduled factor infusions as prophylaxis to prevent long-term joint disease in patients with severe hemophilia.65,66 There is also retrospective evidence to suggest that such scheduled prophylaxis can decrease the risk of ICH in patients with hemophilia.33 Prophylaxis is better established in patients with hemophilia A than in patients with 10 Reprints: www.ebmedicine.net/pempissues hemophilia B, but evidence supports its use in both groups.67 Consequently, many patients with severe hemophilia will be receiving regular factor infusions. A typical prophylaxis schedule is infusion 3 times weekly for factor VIII and twice weekly for factor IX, though a variety of prophylaxis schedules exist. Note, however, that even for patients who are nominally on prophylaxis, compliance can be a significant challenge, especially in adolescent patients, and compliance should not be assumed.68 30 to 60 ristocetin cofactor units/kg of vWF with maintenance dosing of 20 to 40 units/kg every 12 to 48 hours. Trough vWF:RCo and factor VIII levels should be monitored daily and maintained at > 50 IU/dL for 3 to 5 days. For major bleeding, a loading dose of 40 to 60 units/kg of vWF is recommended, followed by a maintenance dose of 20 to 40 units/kg every 8 to 24 hours thereafter. Trough vWF:RCo and factor VIII levels should be monitored daily and maintained at > 50 IU/dL for at least 7 days. Care should be taken to avoid excessively high factor VIII levels, which may occur over time. See Table 5 for a quick reference on vWF dosing recommendations. Treatment Of Bleeding From Von Willebrand Disease Bleeding tends to be less severe from vWD compared to hemophilia, and life-threatening bleeding in most types of vWd is uncommon. However, familiarity with potential interventions is important. Treatment depends on the subtype of vWD and can include: increasing plasma concentration of vWF by stimulation of endogenous vWF release from endothelial cells through administration of desmopressin, replacing vWF using plasma-derived concentrates, and/or using agents that promote hemostasis but do not substantially alter plasma concentrations of vWF (ie, antifibrinolytics). Treatment choice largely depends on whether or not the patient has demonstrated a prior response to desmopressin.1 Adjunctive Therapies Adjunctive therapies are used for specific bleeding types in vWD. For menorrhagia, hormonal contraceptives are first-line therapy, with desmopressin and antifibrinolytics representing adjunctive therapies.15 For mucosal bleeding in both vWD and hemophilia, antifibrinolytic drugs (specifically aminocaproic acid and tranexamic acid) can be useful adjuncts. These medications inhibit the conversion of plasminogen to plasmin, which inhibits fibrinolysis, and stabilizes clots that have already formed. They are a useful adjunct to increase vWF levels in patients with vWD, and they are particularly helpful in managing oral bleeding or after oral surgery, as the oral cavity is high in fibrinolytic activity. Dosing of aminocaproic acid is 50 to 60 mg/kg (max 5 g) intravenously or by mouth every 6 hours until bleeding is controlled, or for 5 to 7 days postoperatively.8 Dosing of tranexamic acid is 10 to 15 mg/kg oral or intravenously every 8 to 12 hours.8,14 The major contraindication to antifibrinolytic use is hematuria, as antifibrinolytic agents can precipitate renovascular thrombi. A urinalysis to exclude blood is recommended prior to initiating an antifibrinolytic.15 Desmopressin Treatment If the patient is known to be responsive to desmopressin, desmopressin can be administered intravenously for minor bleeds at a dose of 0.3 mcg/kg or intranasally at a dose of one 150-mcg spray for patients weighing < 50 kg or 300 mcg (2 sprays) for persons weighing ≥ 50 kg. Of note, desmopressin nasal spray used to treat vWD is a higher concentration (150 mcg/spray) than that used for other indications (such as enuresis), and care should be taken to ensure the proper concentration of solution is administered.69 Side effects of desmopressin include facial swelling, headache, and, most importantly, water retention, as desmopressin mimics the effect of antidiuretic hormone at the kidneys and causes free-water retention. Patients should be instructed to limit fluid intake to maintenance levels and avoid intake of hypotonic fluids for 24 hours following desmopressin administration. If intravenous fluids are administered, normal saline should be used. If multiple doses are required, serum electrolytes should be monitored. To avoid tachyphylaxis, which occurs as endothelial vWF stores become exhausted, discontinue desmopressin after 2 to 3 doses.8 For patients with major bleeding or for patients who are not responsive to desmopressin, vWF-containing concentrates are required. Table 5. Dosing Of Von Willebrand Factor Concentrates For Major And Minor Bleeding And Surgery15 Von Willebrand Factor Concentrates For minor bleeding in patients who are not responsive to desmopressin, administer a loading dose of September 2015 • www.ebmedicine.net Type of Bleeding or Surgery Goal of Treatment (vWF:RCo) Initial IV Dose of vWF Duration of Treatment Major > 50 IU/dL for 7-14 days 40-60 units/kg 7-14 days; administer 20-40 units/kg every 8-24 hours to maintain vWF:RCo > 50 IU/dL Minor > 50 IU/dL for 3-5 days 30-60 units/kg 3-5 days; administer 2040 units/kg every 1248 hours to maintain vWF:RCo > 50 IU/dL Abbreviations: IU, international units; IV, intravenous; vWF, von Willebrand factor; vWF:RCo, von Willebrand factor ristocetin cofactor. 11 Mobile app access: www.ebmedicine.net/app Acute Management Of Common Bleeds In Patients With Hemophilia Patient with a known diagnosis of hemophilia presents to the ED Priority triage: evaluate the patient within 15 minutes of arrival and notify the patient's hematologist as soon as possible Concern for ICH: Patient presents with head trauma, headache, neurologic findings, or signs of increased ICP, with severe hemophilia or inhibitors Hemarthrosis or muscle bleed Administer factor concentrate: • 20-40 units/kg for hemophilia A, 40-60 units/kg for hemophilia B (Class I) • 270 mcg/kg rFVIIa for patient with inhibitors (FDA-approved dose: 90 mcg/kg) (Class I) Administer factor concentrate: • 50 units/kg FVIII for hemophilia A, 100 units/kg FIX for hemophilia B (Class II) • 270 mcg/kg rFVIIa for patient with inhibitors (Class II) • Hemarthrosis: If symptoms are stable and caregivers are comfortable with home monitoring, discharge to home (Class II) • Significant muscle bleeding: Admit for monitoring for compartment syndrome and/or nerve compression (Class II) Obtain head CT Positive for ICH • Admit to the ICU • Obtain a neurosurgical consultation • Maintain factor level > 80% for 7 days, then > 50% for 7 days (Class II) Negative for ICH • Discharge to home if asymptomatic • Consider admission for observation Abbreviations: CT, computed tomography; FDA, United States Food and Drug Administration; FIX, factor IX; FVIII, factor VIII; ICH, intracranial hemorrhage; ICP, intracranial pressure; ICU, intensive care unit; rFVIIa, recombinant factor VIIa. Class Of Evidence Definitions Each action in the clinical pathways section of Pediatric Emergency Medicine Practice receives a score based on the following definitions. Class I • Always acceptable, safe • Definitely useful • Proven in both efficacy and effectiveness Level of Evidence: • One or more large prospective studies are present (with rare exceptions) • High-quality meta-analyses • Study results consistently positive and compelling Class II • Safe, acceptable • Probably useful Level of Evidence: • Generally higher levels of evidence • Nonrandomized or retrospective studies: historic, cohort, or case control studies • Less robust randomized controlled trials • Results consistently positive Class III • May be acceptable • Possibly useful • Considered optional or alternative treatments Level of Evidence: • Generally lower or intermediate levels of evidence • Case series, animal studies, consensus panels • Occasionally positive results Indeterminate • Continuing area of research • No recommendations until further research Level of Evidence: • Evidence not available • Higher studies in progress • Results inconsistent, contradictory • Results not compelling This clinical pathway is intended to supplement, rather than substitute for, professional judgment and may be changed depending upon a patient’s individual needs. Failure to comply with this pathway does not represent a breach of the standard of care. Copyright © 2015 EB Medicine. 1-800-249-5770. No part of this publication may be reproduced in any format without written consent of EB Medicine. Copyright © 2015 EB Medicine. All rights reserved. 12 Reprints: www.ebmedicine.net/pempissues Emergency Department Workup For Suspected Bleeding Disorder Patient presents with a history of significant bleeding Low platelets, PT/PTT abnormal Send screening laboratory tests: CBC, PT, PTT, fibrinogen Low platelets, PT/PTT normal Consider and work up DIC, sepsis, or other significant systemic derangements Consider and work up systemic causes of thrombocytopenia: ITP, HUS, autoimmune disorder, bone marrow disease, etc PT/PTT abnormal, normal CBC PTT only abnormal PTT and PT abnormal PT only abnormal Consider: • FVIII, FIX, FXI deficiencies, vWD • Presence of heparin or lupus anticoagulant Consider: • FV, FX, prothrombin, or fibrinogen deficiencies • Liver disease • Exposure to rat poison • Hemorrhagic disease of the newborn Consider: • FVII deficiency • Warfarin use • Liver disease • Exposure to rat poison • Hemorrhagic disease of the newborn Consult hematology and send: • FVIII, FIX, FXI levels • von Willebrand panel (vWF:Ag, vWF:RCo, FVIII level) • Mixing study to assess for lupus anticoagulant and other inhibitors • Verify no heparin in sample Consult hematology and send: • FV, FX, and FII levels • Fibrinogen level Consult hematology and send: • FVII level • Severity of the bleed should dictate disposition • For patients with abnormal PTT (which suggests a diagnosis of hemophilia) and severe bleeding, admission should be strongly considered PT/PTT normal, normal CBC Consider: • von Willebrand disease • Factor XIII deficiency • Fibrinolytic disorders (rare) • Functional platelet disorder • Connective tissue disorders • Vitamin deficiencies Consult hematology and send: • von Willebrand panel (vWF:Ag, vWF:RCo, FVIII level) • Thrombin time Consider: • Micronutritent/vitamin levels • Consider platelet function assay platelet aggregation studies to assess for functional platelet disorder Abbreviations: CBC, complete blood count; DIC, disseminated intravascular coagulopathy; FII, factor II; FIX, factor IX; FV, factor V; FVII, factor VII; FVIII, factor VIII; FX, factor X; FXI, factor XI; HUS, hemolytic uremic syndrome; ITP, idiopathic thrombocytopenic purpura; PT, prothrombin time; PTT, partial thromboplastin time; vWD, von Willebrand disease; vWF:Ag, von Willebrand factor antigen; vWF:RCo, von Willebrand factor ristocetin cofactor. September 2015 • www.ebmedicine.net 13 Mobile app access: www.ebmedicine.net/app Special Populations with bypassing agents should, ideally, be administered within 1 hour of bleeding onset, and homebased treatment may be appropriate for patients with milder bleeds such as hemarthroses.78-80 Hospitalization and repeated bypassing agent administration is recommended for severe or abnormal bleeds.38,76 There was initially significant concern that the use of rFVIIa could lead to an increased risk of thromboembolic events. However, a 2006 review of 185 thromboembolic events occurring in patients given rFVIIa demonstrated that only 17 events occurred in patients with hemophilia, with the vast majority of thrombi occurring in patients without hemophilia who were receiving rFVIIa off-label. Consequently, rFVIIa is generally considered safe for both pediatric and adult patients with hemophilia.81,82 In addition to its use in acute bleeds, there is also some evidence to support prophylactic use of rFVIIa in patients who have inhibitors and significant bleeding frequency.83 Patients With Hemophilia With Inhibitors Clotting factor inhibitors develop in approximately 33% of patients with hemophilia A and 6.5% of patients with hemophilia B.70,71 Inhibitor development represents the most significant complication of treatment in patients with hemophilia and significantly complicates treatment of acute bleeds. Development of inhibitors is more likely in patients who have specific genotypes of hemophilia, patients who are exposed to factor concentrates at a younger age, and patients who have more exposure to early intensive therapy.72 Inhibitor activity is measured in Bethesda units (BU). Patients with an inhibitor titer < 5 BU/mL are considered low titer, whereas patients with an inhibitor titer ≥ 5 BU/mL are considered high titer. In addition, a patient is considered to have lowresponding inhibitor when the inhibitor titer remains < 5 BU/mL despite repeated factor infusions.73 Patients with low-titer, low-responding inhibitors (< 5 BU/mL) can continue to be treated effectively with clotting factor concentrates for acute bleeds. For such patients, a higher dose of factor is required, and the following formula is used to estimate the amount of factor VIII needed as a loading dose to neutralize the inhibitor: [body weight (kg) × 80 × (1-hematocrit) × antibody titer (BU/mL)] and add an additional 50 units/kg above the calculated loading dose to achieve a measurable factor VIII activity level.73 Once a patient has an inhibitor concentration of ≥ 5 BU/mL, as is the case for most patients with inhibitors, factor replacement will not be effective in treatment of bleeding episodes, and a bypassing agent is required. There are 2 bypassing agents available for these patients: activated recombinant factor VIIa (rFVIIa) and factor eight inhibitor bypassing activity (FEIBA).74 FEIBA contains multiple vitamin-K-dependent clotting factors with some in active forms. Typical dosing for FEIBA is 50 to 75 units/kg for joint bleeds and 100 units/kg for life-threatening or limb-threatening bleeding.73 While repeat doses can be administered every 12 hours, it is important to avoid doses > 200 units/kg/day due to thrombotic risk. Recombinant factor VIIa (eg, NovoSeven®, AryoSeven®) contains activated factor VII, which bypasses the intrinsic clotting pathway to directly stimulate clot formation. The standard dose is 90 mcg/kg every 2 to 3 hours for 3 doses, but some studies support a single higher dose of recombinant factor VIIa of 270 mcg/kg.75-77 If further treatment is required, continued doses of 90 mcg/kg every 2 to 3 hours can be administered,38 but if the 270 mcg/kg dose is used, subsequent doses should be separated by at least 6 hours. As with clotting factor concentrates, treatment Copyright © 2015 EB Medicine. All rights reserved. FEIBA Versus Recombinant Factor VIIa For Refractory Bleeding In Patients With Inhibitors Large randomized trials have demonstrated that FEIBA and rFVIIa are equally efficacious in treating bleeding in patients with hemophilia who have high-titer inhibitors.84 However, some individual patients may respond better to one bypassing agent over another.14 Additionally, rFVIIa may be a preferred choice for patients with factor IX deficiency and inhibitors, as these patients have a high incidence of allergic reactions to exposure to factor IX. Similarly, rFVIIa is preferred for patients with factor VIII deficiency with inhibitors if one is limiting exposure to factor VIII while awaiting a decrease in inhibitor titer. The presence of inhibitors is a risk factor for severe bleeding, and 10% to 20% of bleeding events in patients with hemophilia with inhibitors will not be controlled with a single bypassing agent.85 For persistent bleeds, a strategy of initially increasing the dose and/or frequency of bypassing product followed by switching or alternating bypassing agents is recommended.23,32 In a series of 20 patients, sequential therapy with alternating FEIBA and rFVIIa every 6 hours was found to be effective for difficult-to-treat bleeds and it was deemed safe, as it did not result in thrombosis, thrombocytopenia, or disseminated intravascular coagulation.86 Serial/alternating bypass therapy should be reserved for life-threatening or limb-threatening bleeds, and a pediatric hematologist should be involved in all such cases.74 Patients With Mild Hemophilia A: Treatment With Desmopressin For patients with mild hemophilia A, desmopressin can be used in lieu of factor concentrate to effectively raise the serum concentration of factor VIII by mobiliz14 Reprints: www.ebmedicine.net/pempissues ing intrinsic stores of vWF and factor VIII. Dosing is the same as that for vWD, and desmopressin can be administered intravenously (0.3 mcg/kg) or intranasally (150 mcg for patients weighing < 50 kg, 300 mcg for patients weighing ≥ 50 kg) with excellent effect, raising factor VIII twofold to sixfold from baseline, which is generally adequate for most simple bleeds.87,88 As with vWD, patients with mild hemophilia A should have a desmopressin challenge to ensure that they are responsive to desmopressin therapy before this is used as a first-line treatment.89 In contrast to hemophilia A, there is virtually no evidence to support use of desmopressin in the treatment of mild hemophilia B.90 of a specific protein factor, it is an appealing target for gene therapy, which has shown significant promise in early small trials, though significantly more research is needed in this area.96,97 Disposition Disposition depends on the type and severity of bleeding, and recommended disposition by bleed type is included in Table 4 (see page 9). For patients who will be sent home, family reliability and comfort with monitoring, and, potentially, with home factor infusion, is critical, as is regular communication with a hematologist. A clear point of contact with a hematologist is a critical component of safe discharge. Patients In Resource-Limited Settings While recombinant factor products are clearly the preferred agents, where available, and virally inactivated plasma-derived products are the second choice, in settings where factor VIII or factor IX concentrates are not available and cannot be obtained, cryoprecipitate can be used to treat hemophilia A and fresh-frozen plasma can be used to treat hemophilia B.44,91,92 Cryoprecipitate, which contains high levels of factor VIII and vWF, may be used initially at a dose of 1 bag/6 kg body weight, to a maximum of 10 units, to treat acute bleeding in patients with hemophilia A. For patients with factor IX deficiency in these settings, fresh-frozen plasma may be administered at a dose of 15 mL/kg as an initial dose. For patients with severe vWD in the resource-limited setting, cryoprecipitate can be used at the same dosing noted for hemophilia A.1 In settings where there is some access to factor concentrates, but such access is limited and cost is a major constraint, emergency clinicians can refer to the World Federation of Hemophilia guidelines for more reserved dosing of factor for various types of bleeds.93 Summary Hemophilia and vWD are the most common inherited bleeding disorders. The central tenet of management of bleeding in hemophilia is to administer the appropriate factor concentrate early and at an appropriate dose to achieve hemostasis. Factor VIII should be administered for patients with hemophilia A, factor IX for patients with hemophilia B, and a bypassing agent (FEIBA or rFVIIa) for patients with inhibitors. For vWD, bleeding is typically less severe. If treatment is required, most patients with type 1 vWD will respond to desmopressin, but a demonstrated response to this therapy is necessary before relying on it. Alternatively, a vWF concentrate can be used. Most patients with hemarthrosis can be managed as outpatients after initial assessment. Patients with more-severe hemorrhages should be admitted to the hospital for tight control of factor levels and close monitoring for sequelae of bleeds. Consultation with the patient’s primary hematologist can provide invaluable support in the evaluation and management of patients with established diagnoses. For patients who do not have a known diagnosis of a bleeding disorder and present to the ED with atypical bleeding, PT/PTT/fibrinogen and CBC represent the most important initial screening laboratory tests, and the results of these studies guide more-specific testing and management. Disposition can then be determined in collaboration with a hematologist for inpatient admission versus discharge to home with close hematology clinic follow-up. Controversies And Cutting Edge In the United States, the annual cost of care for patients with hemophilia is high, with a 2015 study estimating the mean cost of care (annually per patient) for patients with severe hemophilia on episodic treatment to be $201,471, and $301,392 for patients on factor prophylaxis. The cost of factor concentrates represent the greatest contributor to this cost.94 For patients on prophylactic factor infusions, frequent self-infusion, up to 4 times weekly, is often required because of the relatively short half-life of factors VIII and IX in circulation. Efforts are underway to synthesize factor fusion proteins and PEGylated factor products with longer serum half-lives, which would require much less frequent dosing and potentially improve compliance and quality of life. Two such products are now approved, Eloctate® (factor VIII) and Alprolix® (factor IX).95 In addition, because hemophilia represents a disease resulting from diminished levels or absence September 2015 • www.ebmedicine.net Case Conclusions You gave the 7-year-old boy 100 units/kg of BeneFIX® factor IX immediately to bring his factor level to 100%. A head CT scan was subsequently obtained demonstrating a small subdural hematoma without overlying skull fracture. He was admitted to the intensive care unit under the neurosurgical service for serial neurological examinations for 2 days, and he continued to receive factor IX infu15 Mobile app access: www.ebmedicine.net/app Risk Management Pitfalls In Hemophilia And Von Willebrand Disease In Children 1. “I wanted to confirm on imaging that an intracranial bleed was actually present before I infused factor.” Factor should be administered immediately upon suspicion of intracranial hemorrhage. Delay in factor administration has the potential to increase morbidity and mortality, and the risk of factor administration is minimal. When in doubt, infuse factor. 7. “This patient with hemophilia did not bring his factor concentrate with him to the ED, and we don’t stock his brand of concentrate here, so we should wait until his factor arrives from home to treat his hemarthrosis.” There is no clear evidence that switching between factor brands increases the risk of inhibitor development. Depending on the severity of the bleed and the anticipated delay in acquiring the patient’s home product, administration of another brand of product may be appropriate. However, recombinant products are preferred, rather than switching from a recombinant product to a plasma-derived product. 2. “This patient had a high-titer inhibitor, so I gave higher doses of factor to overcome the inhibitor to try to stop the bleeding.” High-titer inhibitors (> 5 BU/mL) cannot be overcome with higher doses of factor concentrates. A bypassing agent (FEIBA or rFVIIa) is required to treat bleeding in these patients. 8. “This patient with vWD needs emergent surgery. We can just give desmopressin preoperatively, and that should be fine.” This may be true; however, a documented response to desmopressin with an appropriate increase in vWF is necessary before relying on this therapy. If the patient has not been shown to be responsive to desmopressin (either has not had a test confirming desmopressin response or has had a test which showed nonresponsiveness to desmopressin), a factor product containing large concentrations of vWF is indicated rather than using desmopressin. 3. “This patient with severe hemophilia only had minimal head trauma and has a normal examination. He can’t have a significant ICH.” Most patients with hemophilia with ICH report only minor trauma history, and many will have a completely normal physical examination. 4. “This infant has no family history of hemophilia, so he can’t have hemophilia.” Thirty percent of cases of hemophilia are new mutations without any family history. Maintain a high index of suspicion for hemophilia in a male patient with significant bleeding, and maintain a low threshold for screening PT/PTT. 9. “My patient with hemophilia is complaining of numbness along the lateral thigh and is having trouble extending his right leg. We should get a neurology consultation and head imaging, as this may be a stroke.” These are typical presenting signs of an iliopsoas hemorrhage. Treatment should be centered on factor replacement to 80%, and imaging should be directed at the psoas region by CT, MRI, or ultrasound. Significant blood loss in this area is possible, and transfusion may be required. 5. “We don’t have any factor VIII concentrates available here. There is nothing I can do for this patient with hemophilia A and worsening hemarthrosis.” In settings where factor VIII concentrate is unavailable, cryoprecipitate (which contains high levels of factor VIII) may be used at a dose of 1 bag/6 kg body weight, to a max of 10 bags, initially to treat acute bleeding. 10. “This patient has menorrhagia and easy bruising, and her mother and sister have a history of similar symptoms. She already had a normal von Willebrand panel test, so she can’t have vWD.” Many factors affect measured vWF levels, including stress, illness, exercise, reproductive hormone levels, and specimen storage and transport. False negatives are common, and a single negative test does not rule out the disease, especially in patients with signs and symptoms suggestive of the disease. 6. “This patient’s factor VIII level is low. He must have hemophilia A.” This is likely the case, but he may also have type 2N or type 3 vWD. Distinguishing between these diseases will help guide factor replacement, and a von Willebrand panel should be sent. Copyright © 2015 EB Medicine. All rights reserved. 16 Reprints: www.ebmedicine.net/pempissues sions of 100 units/kg for 7 days, after which he was given factor infusions of 50 units/kg for 7 days before resuming his normal prophylaxis regimen. He had no long-term neurologic sequelae. You gave the 17-year-old boy a single dose of 270 mcg/kg of rFVIIa (NovoSeven®). A compression bandage was applied to the affected knee, and he was discharged to home from the ED with hematology clinic follow-up the next day. At follow-up, his swelling was starting to improve, so no further rFVIIa was given. Within 1 week, swelling and function had improved. The 16-year-old patient with menorrhagia was told that she likely had von Willebrand disease, and arrangements were made for a desmopressin challenge test in the hematology clinic. After discussion with the patient and her family and review for contraindications to oral contraceptive pills, the patient was started on an oral contraceptive pill, and her periods subsequently normalized within 2 months. 1.* Singleton T, Kruse-Jarres R, Leissinger C. Emergency department care for patients with hemophilia and von Willebrand disease. J Emerg Med. 2010;39(2):158-165. (Review) 2.* Josephson N. The hemophilias and their clinical management. Hematology Am Soc Hematol Educ Program. 2013;2013:261-267. (Review) Di Michele D, Neufeld EJ. Hemophilia: a new approach to an old disease. Hematol Oncol Clin North Am. 1998;12(6):13151344. (Review) 4. Mannucci PM, Mancuso ME, Santagostino E. How we choose factor VIII to treat hemophilia. Blood. 2012;119(18):4108-4114. (Review) 5. Lovdahl S, Henriksson KM, Baghaei F, et al. Incidence, mortality rates and causes of deaths in haemophilia patients in Sweden. Haemophilia. 2013;19(3):362-369. (Retrospective cohort study; 1431 cases and 7150 controls) 6.* Rodeghiero F, Castaman G, Tosetto A. How I treat von Willebrand disease. Blood. 2009;114(6):1158-1165. (Expert opinion with literature review) 7.* Yawn B, Nichols WL, Rick, ME. Diagnosis and management of von Willebrand disease: guidelines for primary care. Am Fam Phys. 2009;80(11):1261-1268. (Expert consensus guidelines) References 8.* Mannucci PM. Treatment of von Willebrand’s disease. N Engl J Med. 2004;351(7):683-694. (Review) Evidence-based medicine requires a critical appraisal of the literature based upon study methodology and number of subjects. Not all references are equally robust. The findings of a large, prospective, randomized, and blinded trial should carry more weight than a case report. To help the reader judge the strength of each reference, pertinent information about the study, such as the type of study and the number of patients in the study will be included in bold type following the references, where available. The most informative references cited in this paper, as determined by the author, will be noted by an asterisk (*) next to the number of the reference. 9. Acharya SS. Rare bleeding disorders in children: identification and primary care management. Pediatrics. 2013;132(5):882-892. (Review) 10. Nuss R, Hoffman R, Hammond L. ED Visits by males with hemophilia. Am J Emerg Med. 2002;20(2):74-78. (Medical record review; 51 patients) 11. Ozgonenel B, Zia A, Callaghan MU, et al. Emergency department visits in children with hemophilia. Pediatr Blood Cancer. 2013;60(7):1188-1191. (Medical record review; 536 emergency department visits, 84 male patients) 12. Minhas HL, Giangrande PL. Presentation of severe haemophilia--a role for accident and emergency doctors? Emerg Med J. 2001;18(4):246-249. (Questionnaire and survey; 25 patients) 13. Richards M, Williams M, Chalmers E, et al. A United Kingdom Haemophilia Centre Doctors’ Organization guideline approved by the British Committee for Standards in Haematology: guideline on the use of prophylactic factor VIII concentrate in children and adults with severe haemophilia A. Br J Haematol. 2010;149(4):498-507. (Practice guidelines) Time- And Cost-Effective Strategies 14.* Treatment Guidelines Working Group. Guidelines for the management of hemophilia. 2nd ed. Montreal, Quebec: World Federation of Hemophilia. 2012. (Consensus guidelines) • Imaging is generally unnecessary in patients with typical signs and symptoms of hemarthrosis, and, in general, hemarthrosis can be treated at home rather than in the hospital. The caveat to this recommendation is that caregivers of patients who have not had prior significant hemarthroses may be less comfortable with home management, and the patients should, therefore, be monitored as inpatients. • Early factor replacement helps decrease total factor requirement and length of hospitalization, and, therefore, the total cost in all bleed types. • Interpret vWD panels with caution in patients with acute illness. Von Willebrand factor levels are elevated during times of stress, and laboratory tests drawn in such settings can be difficult to interpret, possibly resulting in false negatives. September 2015 • www.ebmedicine.net 3. 15.* Nichols WL, Hultin, MB, James AH, et al. The diagnosis, evaluation and management of von Willebrand disease. National Heart Lung and Blood Institute. NIH Publication No. 08-5832. Bethesda, MD; 2012. (Practice guidelines) 16.* Fowler H, Lacey R, Keaney J, et al. Emergency and out of hours care of patients with inherited bleeding disorders. Haemophilia. 2012;18(3):e126-e131. (Practice guidelines audit) 17. Key NS, Negrier C. Coagulation factor concentrates: past, present and future. Lancet. 2007;370(9585):439-448. (Review) 18. Rosenberg PS, Goedert JJ. Estimating the cumulative incidence of HIV infection among persons with haemophilia in the United States of America. Stat Med. 1998;17(2):155-168. (Multicenter data analysis) 19. Darby SC, Kan SW, Spooner RJ, et al. Mortality rates, life expectancy, and causes of death in people with hemophilia A or B in the United Kingdom who were not infected with HIV. Blood. 2007;110(3):815-825. (Retrospective review; 6018 cases) 17 Mobile app access: www.ebmedicine.net/app 20. Kumar R, Carcao. Inherited abnormalities of coagulation: hemophilia, von Willebrand disease, and beyond. Pediatr Clin North Am. 2013;60(6):1419-1441. (Review) gies for securing and maintaining predictable efficacy with recombinant activated factor VII. Haemophilia. 2012;18(2):255262. (Expert consensus recommendations) 21. Robertson J, Lillicrap D, James PD. von Willebrand disease. Pediatr Clin North Am. 2008;55(2):377-392. (Review) 22. Mortimore W. Focus on diagnosis: screening for von Willebrand disease. Pediatr Rev. 2005;26(302):301-303. (Expert opinion and review) 39. Blankenship CS, Tortella BJ, Bruno, M. BE EMPOWERED, a specialty pharmacy education program for hemophilia B patients, impacts adult joint bleeds and pediatric use of RICE. J Manag Care Pharm. 2014;20(2):151-158. (Prospective cohort study; 21 subjects) 23. Zimmerman B, Valentino LA. Hemophilia: in review. Pediatr Rev. 2013;34(7):289-295. (Review) 40. Gill JC. Therapy of factor VIII deficiency. Semin Thromb Hemost. 1993;19(1):1-12. (Review) 24. Di Michele DM, Gibb C, Lefkowitz JM, et al. Severe and moderate haemophilia A and B in US females. Haemophilia. 2014;20(2):e136-e143. (Multicenter retrospective study; 22 patients) 41. Nagel K, Pai MK, Paes BA, et al. Diagnosis and treatment of intracranial hemorrhage in children with hemophilia. Blood Coagul Fibrinolysis. 2013;24(1):23-27. (Review) 25. Sharathkumar A, Pipe S. Bleeding disorders. Pediatr Rev. 2008;29(4):121-130. (Review) 42. Witmer CM, Raffini LJ, Manno CS. Utility of computed tomography of the head following head trauma in boys wth haemophilia. Haemophilia. 2007;13(5):560-566. (Review) 26. Pollmann H, Richter H, Ringkamp H, et al. When are children diagnosed as having severe haemophilia and when do they start to bleed? A 10-year single-centre PUP study. Eur J Pediatr. 1999;158 Suppl 3:S166-S170. (Prospective singlecenter study; 37 patients) 43. Traivaree C, Blanchette V, Armstrong G, et al. Intracranial bleeding in haemophilia beyond the neonatal period - the role of CT imaging in suspected intracranial bleeding. Haemophilia. 2007;13(5):552-559. (Retrospective chart review; 43 patients) 27. Venkateswaran L, Wilimas JA, Jones DJ, et al. Mild hemophilia in children: prevalence, complications and treatment. J Pediatr Hematol Oncol. 1998;20(1):32-35. (Retrospective chart review; 55 patients) 44. Hermans C, De Moerloose P, Fisher K, et al. Management of acute haemarthrosis in haemophilia A without inhibitors: literature review, European survey and recommendations. Haemophilia. 2011;17(3):383-392. (Review) 28. Berntorp E, Halimeh S, Gringeri A, et al. Management of bleeding disorders in children. Haemophilia. 2012;18 Suppl 2:15-23. (Review) 45. Melchiorre D, Linari S, Innocenti M, et al. Ultrasound detects joint damage and bleeding in haemophilic arthropathy: a proposal of a score. Haemophilia. 2011;17(1):112-117. (Prospective cohort study; 83 joint bleeds) 29. Nolan B, Vidler V, Vora A, et al. Unsuspected haemophilia in children with a single swollen joint. BMJ. 2003;326(7381):151152. (Case reports; 2 patients) 46. Sorensen B, Benson GM, Bladen M, et al. Management of muscle haematomas in patients with severe haemophilia in an evidence poor world. Haemophilia. 2012;18(4):598-606. (Review) 30. Beyer R, Ingersley J, Sorensen B. Muscle bleeds in professional athletes - diagnosis, classification, treatment and potential impact in patients with haemophilia. Haemophilia. 2010;16(6):858-865. (Review paper) 47. Matysiak M, Bobrowska H, Balwierz W, et al. Clinical experience with Optivate®, high-purity factor VIII (FVIII) product with von Willebrand factor (VWF) in young children with haemophilia A. Haemophilia. 2011;17(5):737-742. (Prospective cohort study; 25 children) 31. Plummer ES, Crary SE, Buchanan GR. Prominent forehead hematomas (“goose-eggs”) as an initial manifestation of hemophilia. J Pediatr. 2013;163(6):1781-1783. (Retrospective chart review; 324 patients) 48. Pollmann H, Externest D, Ganser A, et al. Efficacy, safety and tolerability of recombinant factor VIII (REFACTO) in patients with haemophilia A: interim data from a postmarketing surveillance study in Germany and Austria. Haemophilia. 2007;13(2):131-143. (Postmarketing surveillance study, 217 patients) 32. Teitel J, Berntorp E, Collins P, et al. A systematic approach to controlling problem bleeds in patients with severe congenital hemophilia A and high-titre inhibitors. Haemophilia. 2007;13(3):256-263. (Expert panel consensus guidelines) 33. Witmer C, Presley R, Kulkarni J, et al. Associations between intracranial haemorrhage and prescribed prophylaxis in a large cohort of haemophilia patients in the United States. Br J Haematol. 2010;152(2):211-216. (Case-control study; 199 patients) 49. Scharrar I, Ehrlich HJ. Lack of evidence for increased inhibitor incidence in patients switched from plasma-derived to recombinant factor VIII. Haemophilia. 2001;7(4):346-348. (Review) 50. Rubinger M, Lillicrap D, Rivard GE, et al. A prospective surveillance study of factor VIII inhibitor development in the Canadian haemophilia A population following the switch to a recombinant factor VIII product formulated with sucrose. Haemophilia. 2008;14(2):281-286. (Retrospective cohort study) 34. Labarque V, Stain AM, Blanchette V, et al. Intracranial haemorrhage in von Willebrand disease: a report on six cases. Heamophilia. 2013;19(4):602-606. (Retrospective chart review; 24 patients) 35. Antunes SV, Vicari P, Cavalheiro S, et al. Intracranial haemorrhage among a population of haemophilic patients in Brazil. Haemophilia. 2003;9(5):573-577. (Retrospective cohort study; 401 patients, 45 with intracranial hemorrhage) 51. Mannucci PM. Treatment of haemophilia: building on strength in the third millenium. Haemophilia. 2011;17(Suppl 3):1-24. (Review) 52. Branchford BR, Monahan PE, Di Paola J. New developments in the treatment of pediatric hemophilia and bleeding disorders. Curr Opin Pediatr. 2013;25(1):23-30. (Review) 36. Sun GH, Aauger KA, Aliu O, et al. Posttonsillectomy hemorrhage in children with von Willebrand disease in hemophilia. JAMA Otolaryngol Head Neck Surg. 2013;139(3):245-249. (Retrospective chart review; 508 patients) 53. Nguyen DD, Takenaka K. Evaluation and management of hereditary hemophilia in the emergency department. J Emerg Nurs. 2009;35(5):437-441. (Review) 37. Kavakli K, Yesilipek A, Antmen B, et al. The value of early treatment in patients with haemophilia and inhibitors. Haemophilia. 2010;16(3):487-494. (Retrospective cohort study; 129 bleeding episodes) 54. Alexander M, Barnes C, Barnett P. Prospective audit of patients with haemophilia: Bleeding episodes and management. J Pediatr and Child Health. 2012;48(2):177-179. (Prospective cohort study; 66 children) 38. Sørensen B, Dargaud Y, Kenet G, et al. On-demand treatment of bleeds in haemophilia patients with inhibitors; strate- Copyright © 2015 EB Medicine. All rights reserved. 18 Reprints: www.ebmedicine.net/pempissues 55. Petrini P. Treatment strategies in children with hemophilia. Pediatr Drugs. 2002;4(7):427-437. (Review) patient with a factor VIII inhibitor. Blood. 2009;113(1):11-17. (Review) 56. Kisker CT, Burke C. Double-blind studies on the use of steroids in the treatment of acute hemarthrosis in patients with hemophilia N Engl J Med. 1970;282(12):639-642. (Review) 74. Collins PW, Chalmers E, UK Hemophilia Centre Doctors, et al. Diagnosis and treatment of factor VIII and IX inhibitors in congenital haemophilia: (4th edition). UK Haemophilia Centre Doctors Organization. Br J Haematol. 2013;160(2):153170. (Practice guidelines) 57. Rodriguez-Merchan E. Acute compartment syndrome in haemophilia. Blood Coagul Fibrinolysis. 2013;24(7):677-682. (Systematic review; 34 patients) 75. Pan-Petesch B, Laguna P, Mital A, et al. Single-dose (270 ug/ kg) recombinant activated factor VII for the treatment and prevention of bleeds in haemophilia A patients with inhbitors: experience from seven European haemophila centers. Haemophilia. 2009;15(3):760-765. (Retrospective study and expert recommendation; 8 patients) 58. Ashrani AA, Osip J, Christie B, et al. Iliopsoas haemorrhage in patients with bleeding disorders--experience from one centre. Haemophilia. 2003;9(6):721-726. (Retrospective review; 297 patients) 59. Balkan C, Kavakli K, Karapinar D. Iliopsoas haemorrhage in patients with haemophilia: results from one centre. Haemophilia. 2005;11(5):463-467. (Chart review; 146 patients) 76. Santagostino E, Mancuso ME, Rocino A, et al. A prospective randomized trial of high and standard dosages of recombinant factor VIIa for treatment of hemarthroses in hemophiliacs with inhibitors. J Thromb Haemost. 2006;4(2):367-371. (Randomized trial; 20 patients) 60. Fernandez-Palazzi F, Hernandez SR, De Bosch NB, et al. Hematomas within the iliopsoas muscles in hemophilic patients: the Latin American experience. Clin Orthop. 1996;328:19-24. (Retrospective cohort study) 77. Laguna P, Mital A. Single higher dose of recombinant activated factor VII in the treatment of hemorrhages in patients with hemophilia complicated by inhibitors. Adv Clin Exp Med. 2012;21(4):519-524. (Prospective trial; 7 patients) 61. Revel-Vilk S, Golomb MR, Achonu C, et al. Effect of intracranial bleeds on the health and quality of life of boys with hemophilia. J Pediatr. 2004;144(4):490-495. (Case-control study; 172 patients, 18 with intracranial hemorrhage) 78. Parameswaran R, Shapiro AD, HTRS Registry Investigators, et al. Dose effect and efficacy of rFVIIa in the treatment of haemophilia patients with inhbitors: analysis from the Hemophilia and Thrombosis Research Society Registry. Haemophilia. 2005;11(2):100-106. (Retrospective chart review; 38 patients, 555 bleeding episodes) 62. Klinge J, Auberger K, Auerswald G, et al. Prevalence and outcome of intracranial haemorrhage in haemophiliacs - a survey of the paediatric group of the German Society of Thrombosis and Haemostasis (GTH). Eur J Pediatr. 1999;158(Suppl 3):S162-S165. (Retrospectve cohort study; 744 patients, 30 intracranial hemorrhages) 79. Young G, Shapiro AD, Walsh CE, et al. Patient/caregiverreported recombinant factor VIIa(rFVIIa) dosing: home treatment of acute bleeds in the Dosing Observational Study in Hemophilia (DOSE). Haemophilia. 2012;18(3):392-399. (Prospective observational study; 158 bleeding episodes) 63. Andes WA, Wulff K, Smith WB. Head trauma in hemophilia. A prospective study. Arch Intern Med. 1984;144(10):1981-1983. (Prospective study; 140 patients) 64. Malec LM, Sidonio RF Jr, Smith KJ, et al. Three costutility analyses of screening for intracranial hemorrhage in neonates with hemophilia. J Pediatr Hematol Oncol. 2014;36(6):474-479. (Cost-utility analysis) 80. Holme PA, Glomstein A, Grønhaug S, et al. Home treatment with bypassing products in inhitor patients: a 7.5 year experience. Haemophilia. 2009;15(3):727-732. (Prospective observational study; 10 patients) 65. Iorio A, Marchesini E, Marcucci M, et al. Clotting factor concentrates given to prevent bleeding and bleeding related complications in people with hemophilia A. Cochrane Database Syst Rev. 2011;9:CD003429. (Systematic review) 81. O’Connell KA, Wood JJ, Wise RP, et al. Thromboembolic adverse events after use of recombinant human coagulation factor VIIa. JAMA. 2006;295(2):293-298. (Database review; 431 adverse event reports) 66. Manco-Johnson MJ, Abshire TC, Shaprio AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357(6):535544. (Randomized controlled trial; 65 patients) 82. Neufeld EJ, Saxena K, Kessler CM, et al. Dosing, efficacy, and safety of recombinant factor VIIa (rFVIIa) in pediatric versus adult patients: the experience of the Hemostasis and Thrombosis Research Society (HTRS) Registry (2004-2008). Pediatr Blood Cancer. 2013;60:1178-1183. (Retrospective database review; 284 children with 1712 episodes, 146 adults with 329 episodes) 67. Nagel K, Walker I, Decker K, et al. Comparing bleed frequency and factor concentrate use between haemophilia A and B patients. Haemophilia. 2011;17(6):872-874. (Data analysis; 78 patients) 83. Konkle BA, Ebbeson LS, Erhardtsen E, et al. Randomized, prospective clinical trial of recombinant factor VIIa for secondary prophylaxis in hemophilia patients with inhibitors. J Thromb Haemost. 2007;5(9):1904-1913. (Randomized prospective trial; 38 patients) 68. Pasi KJ. Haemophila and adolescence. Haemophilia. 2011;17(Suppl. 3):1-24. (Review) 69. Khair K, Baker K, Mathias M, et al. Intranasal desmopressin (Octim): a safe and efficacious treatment option for children with bleeding disorders. Haemophilia. 2007;13(5):548-551. (Prospective noncontrolled single-arm medication trial; 20 children) 84. Astermark J, Donfield, SM, Di Michele DM, et al. A randomized comparison of bypassing agents in hemophilia complicated by an inhibitor: the FEIBA NovoSeven Comparative (FENOC) Study. Blood. 2007;109(2):546-551. (Randomized trial; 96 bleeding episodes in 48 patients) 70. Di Michele D. Inhibitor treatment in haemophilias A and B: inhibitor diagnosis. Haemophilia. 2006;12(Suppl 6):37-42. (Review) 85. Gringeri A, Fischer K, Karafoulidou A, et al. Sequential combined bypassing therapy is safe and effective in the treatment of unresponsive bleeding in adults and children with haemophilia and inhibitors. Haemophilia. 2011;17(4):630-635. (Retrospective database review; 11 patients) 71. Oren H, Yaprak I, Irken G. Factor VIII inhibitors in patients with hemophilia A. Acta Haematologica. 1999;102(1):42-46. (Retrospectve chart review; 58 patients) 72. Hay CR, Palmer B, Chalmers E, et al. Incidence of factor VIII inhibitors throughout life in severe hemophilia A in the United Kingdom. Blood. 2011;117(23):6367-6370. (Database review; 2528 patients) 86. Schneiderman J, Nugent DJ, Young G. Sequential therapy with activated prothrombin complex concentrate and recombinant factor VIIa in patients with severe haemophilia and inhibitors. Haemophilia. 2004;10(4):347-351. (Retrospective 73. Kempton CL, White GC. How we treat a hemophilia A September 2015 • www.ebmedicine.net 19 Mobile app access: www.ebmedicine.net/app 1. Which of the following statements about vWD is TRUE? a. Most cases of vWD show X-linked inheritance. b. Von Willebrand disease always represents a quantitative deficiency in vWF. c. Up to 1% of the United States population meets laboratory criteria for diagnosis of vWD, but far fewer than this have symptomatic disease. d. Most patients with vWD have bleeding episodes similar to those seen in severe hemophilia. chart review; 20 patients) 87. Gill JC, Ottum M, Schwartz B. Evaluation of high concentration intranasal and intravenous desmopressin in pediatric patients with mild hemophilia A or mild-to-moderate type 1 von Willedbrand disease. J Pediatr. 2002;140(5):595-599. (Open-label single-dose trial; 25 patients) 88. Santagostino E. Managing mild haemophila A. Haemophilia. 2011;17(Suppl. 3):21-24. (Review) 89. Revel-Vilk S, Blanchette VS, Sparling C, et al. DDAVP challenge tests in boys with mild/moderate haemophilia A. Br J Haematol. 2001;117(4):947-951. (Retrospective chart review; 62 patients) 90. Ehl S, Severin T, Sutor AH. DDAVP (desmopressin; 1-deamino-cys-8-D-arginine-vasopressin) treatment in children with hemophilia B. Br J Haematol. 2000;111(4):1260-1262. (Prospective trial; 4 patients.) 2. Which of the following statements about hemophilia is TRUE? a. Hemophilia A is the most common inherited bleeding disorder in the United States. b. De novo mutations resulting in hemophilia are extremely rare (< 1%). In the vast majority of cases, a family history of hemophilia can be elicited. c. Hemophilia causes isolated prolongation of the measured PT. d. Patients classified as having severe hemophilia have < 1% of normal clotting factor level activity. 91. Chuansumrit A. Treatment of haemophilia in the developing countries. Haemophilia. 2003;9(4):387-390. (Review) 92. De Kleijn P, Odent T, Berntorp E, et al. Differences between developed and developing countries in paediatric care in haemophilia. Haemophilia. 2012;18 Suppl 4:94-100. (Review) 93. Treatment Guidelines Working Group. Guidelines for the management of hemophilia. Montreal, Quebec: World Federation of Hemophilia. 2005. (Consensus guidelines) 94. Zhou ZY, Koerper MA, Johnson KA, et al. Burden of illness: direct and indirect costs among persons with hemophilia A in the United States. J Med Econ. 2015;18(6):457-465. (Multicenter retrospective review and data analysis; 222 patients) 95. Powell JS, Pasi KJ, Ragni MV, et al. Phase 3 study of recombinant factor IX Fc fusion protein in hemophilia B. N Engl J Med. 2013;369(24):2313-2323. (Phase 3 nonrandomized openlabel study; 123 patients) 3. A 3-year-old boy with no known history or family history of a bleeding disorder presents with a large left ankle hemarthrosis after mild trauma. What are the initial considerations when evaluating this patient? a. This patient cannot have hemophilia given the absence of a history of this disease. b. This patient cannot have hemophilia, as he is already aged 3 years and has had no prior bleeding episodes. c. Screening CBC, PT/PTT/fibrinogen should be sent to evaluate for a bleeding disorder. d. Factors VIII, IX, XI, and XII levels along with a vWD panel and thrombin time should be sent on first screening in the ED. 96. Sharma A, Easow Mathew M, Sriganesh V, et al. Gene therapy for haemophilia. Cochrane Database Syst Rev. 2014;11:CD010822. (Systematic review) 97. Nathwani AC, Tuddenham EG, Rangarajan S, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365(25):2357-2365. (Prospective cohort study; 6 participants) CME Questions Take This Test Online! Current subscribers receive CME credit absolutely free by completing the following test. Each issue includes 4 AMA PRA Category 1 CreditsTM, 4 ACEP Category I credits, 4 AAP Prescribed credits, and 4 Take This Test Online! AOA Category 2A or 2B credits. Monthly online testing is now available for current and archived issues. To receive your free CME credits for this issue, scan the QR code below with your smartphone or visit www.ebmedicine.net/P0915. Copyright © 2015 EB Medicine. All rights reserved. 4. A 7-year-old boy with severe hemophilia A without inhibitors presents to the ED with a clinical examination that is consistent with a right knee hemarthrosis. What is the appropriate management of this patient? a. A single dose of factor VIII concentrate at 20 to 40 units/kg with repeat doses at 12-hour to 24-hour intervals until symptoms have resolved. b. Pain control with ibuprofen and opioids. c. Perform a CT or MRI of the knee to confirm the diagnosis. d. Aspirate the knee joint to confirm the diagnosis. 20 Reprints: www.ebmedicine.net/pempissues 5. A 16-year-old boy with hemophilia A who is on 3-times-weekly prophylaxis presents to the ED with thigh and hip pain and a flexion contracture at the right hip that started while he was sitting in class at school today. He has paresthesias in the femoral nerve distribution on the affected side. Which of the following should be considered in this case? a. Imaging by CT or MRI is not helpful in confirming the diagnosis or following for subsequent resolution. b. Major, life-threatening bleeding may result from this type of bleed and hemoglobin/ hematocrit levels should be checked and serially followed, if decreased. c. The patient should be encouraged to ambulate immediately to improve range of motion in the affected muscle. d. Factor replacement should be dosed at the same level as for hemarthrosis. 8. A 9-year-old girl weighing 28 kg with a history of frequent nosebleeds underwent evaluation by a hematologist. She was found to have laboratory tests consistent with type I vWD, and was also found to be responsive to desmopressin. She is now complaining of worsening epistaxis. Which of the following is a reasonable therapeutic recommendation? a. Administer an intravenous vWF concentrate at an initial dose of 40 to 60 units/kg. b. Administer intranasal desmopressin at a dose of 150 mcg. She should be told to drink plenty of water in order to stay well hydrated. c. Administer intranasal desmopressin at a dose of 300 mcg and restrict fluid intake in the 24 hours following treatment. d. Administer intranasal desmopressin at a dose of 150 mcg and restrict fluid intake to maintenance levels for 24 hours following treatment. 6. A 13-year-old boy with hemophilia B without inhibitors presents to the ED after being struck in the head by a baseball at high speed. He was not wearing a helmet at the time of the incident. He is on prophylaxis twice a week, and he received his last dose of factor 3 days ago. He is presently awake and alert. The first step in initial management in the ED for this patient is: a. Immediate administration of factor IX concentrate at a dose of 50 units/kg b. CT scan of the head as soon as available, followed by factor administration if an intracranial hemorrhage is evident on CT c. Immediate administration of factor IX concentrate at a dose of 100 units/kg d. Administration of rFVIIa at a dose of 270 mcg/kg 9. A 17-year-old girl with type 2 vWD that is not responsive to desmopressin presents to the ED with appendicitis. She requires open appendectomy. What is the appropriate preoperative management of this patient? a. Administer 300 mcg of desmopressin via the intranasal route. b. Administer 0.3 mcg/kg of desmopressin via the intravenous route. c. Administer vWF-containing concentrate 40 to 60 ristocetin cofactor units/kg via the intravenous route. d. Administer factor IX 40 to 60 units/kg via the intravenous route. 10. A 15-year-old boy with hemophilia A and a high-titer inhibitor presents to the ED with a large intramuscular bleed into his left calf. Appropriate management of this patient includes: a. Administration of factor VIII concentrate at a dose of 20 to 40 units/kg, followed by hospital admission for monitoring and repeat administrations, as needed b. Administration of factor VIII concentrate at a dose of 100 units/kg, followed by discharge to home with hematology clinic follow-up within 1 week c. Administration of either FEIBA 50 to 75 units/kg or rFVIIa (90 mcg/kg every 2-3 hours for 3 doses, or a single dose of 270 mcg/kg), followed by hospital admission for monitoring and repeat administrations as needed d. Administration of either intravenous FEIBA 50 to 75 units/kg or rFVIIa 270 mcg/kg followed by discharge to home with hematology follow-up within 1 week 7. Which of the following is a contraindication to adjunctive use of antifibrinolytics in patients with bleeding disorders? a. Bleeding localized to a mucosal site (eg, dental bleeding) b. An underlying diagnosis of type 1 vWD with heavy menses that is not responsive to oral contraceptive pills c. The presence of hematuria d. An underlying diagnosis of mild hemophilia September 2015 • www.ebmedicine.net 21 Mobile app access: www.ebmedicine.net/app Upcoming Issues In Pediatric Emergency Medicine Practice Acute Management Of Inhaled Foreign Bodies In Pediatric Patients In The Emergency Department Use Of Diagnostic Ultrasound In The Emergency Department To Assess Conditions In Pediatric Patients While much is known about the management of inhaled foreign bodies, it remains a significant risk to young children, affecting thousands every year. There is a substantial amount of literature on the topic in otolaryngology and surgery; however, there is limited emergency medicine literature addressing inhaled foreign bodies. This review discusses the etiology, pathophysiology, diagnosis, and management of inhaled foreign bodies. For the purposes of this review, inhaled foreign bodies will refer to foreign bodies (both organic and inorganic) located in the posterior nasopharynx, larynx, trachea, and bronchi. The focus will be on risk factors and clinical clues to the diagnosis, as well as emergent management of inhaled foreign bodies. Performing a diagnostic ultrasound at the point of care in the emergency department can answer focused clinical questions in a rapid manner. Over the last 20 years, the use of ultrasound in the emergency department has become a core requirement in emergency medicine residencies and some pediatric emergency medicine fellowships. In the pediatric setting, the growth has been slower, but there is increasing demand for these studies, given the absence of ionizing radiation with ultrasound. This review focuses on the current evidence for the most common indications for diagnostic ultrasound. Evidence in the pediatric setting is presented, or extrapolated from adult literature where pediatric evidence is scarce. The limitations of diagnostic ultrasound in the emergency department as well as current trends, controversies, and future directions are discussed. Time- And Cost-Effective Strategies • Chest radiography is the first line of investigation in the management of a suspected inhaled foreign body. Normal chest radiography does not exclude the diagnosis of an inhaled foreign body, and thus, additional investigations are sometimes necessary. • Rigid bronchoscopy is considered the standard of care in most centers when evaluating a child with possible foreign body inhalation. This procedure allows for better visualization of the airways, removal of the foreign body with a variety of instruments, and better control of bleeding after removal. Flexible bronchoscopy is routinely used to evaluate a child with recurrent pneumonia or chronic cough, but recently has been shown to be effective and safer than rigid bronchoscopy for excluding the presence of a foreign body in patients with low suspicion of foreign body inhalation. • The American Academy of Pediatrics recommends that anticipatory guidance be provided to parents when their child is 6 months of age. At this age, children begin to develop the fine motor skills needed to pick up small objects. It is important to advise parents not to offer small food items, such as peanuts, until the child is old enough to chew properly. It is also important to advise parents to discourage their child from eating while running, laughing, or playing, and to encourage their child to sit upright. Copyright © 2015 EB Medicine. All rights reserved. Risk Management Pitfalls In The Use Of Ultrasound For Diagnostic Purposes In The Emergency Department 1. “While caring for a child with possible intussusception, I couldn’t obtain a good view of the abdomen on ultrasound, so I figured it was not intussusception, and I discharged the patient.” Emergency ultrasound is meant to answer yes or no questions. If your examination is technically inadequate or you are unsure that you adequately answered your clinical question based on your images, then ask for a radiology-performed ultrasound or another available imaging study. 2. 3. 22 “In a pediatric patient with cardiac arrest, there was no evidence of cardiac activity on ultrasound, so I recommended that we should discontinue resuscitation efforts.” While there are data on adults that ultrasound can be used as a prognostic indicator in cardiac arrest, there are insufficient data in children for it be used alone to prognosticate outcomes in pediatric cardiac arrest. “The patient had a pericardial effusion and was tachycardic. However, I saw no signs of cardiac tamponade on ultrasound, so I did not consult cardiology or cardiac surgery.” Cardiac tamponade is a clinical diagnosis. If the patient has a pericardial effusion and is unstable, then cardiac tamponade should be considered despite the lack of ultrasound findings of tamponade. Reprints: www.ebmedicine.net/pempissues Now Available! Emergency Trauma Care: Current Topics And Controversies, Volume I Writtenbytopemergencyphysiciansandtraumaspecialists from around the country, Emergency Trauma Care: Current Topics And Controversies, Volume I gives you an easy and affordablewaytoearntraumacredits.You’lllearnabout6 ofthemostpressingconcernsfacingemergencyclinicians today,including: • GuidelinesInTrauma–areviewoftheprimary guidelinesforheadandnecktrauma,thoracictrauma, andabdominaltrauma • AirwayManagementInTrauma–themostimportant conceptsandpracticesinsuccessfulairway management • ResuscitationInTrauma–areviewof currentpracticeinmanagingthedirectand indirecteffectsofcoagulopathyintrauma ONLY $189 • TheInitialApproachToPatientsWithModerate ToSevereTraumaticBrainInjury–criticalactionstotakeintheinitial managementofpatientswithheadtrauma • LimitingRadiationExposureInTraumaImaging–bestpracticesandthelatestevidence onhowtoreduceradiationexposurewhilemaximizingdiagnosticeffectiveness • PediatricTraumaUpdate–anorgan-specificapproachtotreatingpediatrictrauma patients 2 EASY WAYS TO ORDER 1. Goonlineto:www.ebmedicine.net/NHBID 2. Call1-800-249-5770or678-366-7933 Use Promotion Code: NHBID at checkout to secure your discount September 2015 • www.ebmedicine.net 23 Mobile app access: www.ebmedicine.net/app Physician CME Information Date of Original Release: September 1, 2015. Date of most recent review: August 15, 2015. Termination date: September 1, 2018. SPECIAL ANNOUNCEMENT The New Pediatric Emergency Medicine Practice Mobile App— Now Available! Mobile-optimized content from Pediatric Emergency Medicine Practice, powered by AgileMD, will enable you to instantly search our entire publication library—so you have access to hundreds of topics on your mobile device at the point of care. Content will be updated in real time so that new clinical topics are available as soon as they are released. The new app also features easy-to-use reference tools, such as bookmarking and note-taking, that enable you to easily follow up on cases and finish research. We are excited to bring you this new subscription benefit—at no charge to you. To download the app, go to www.ebmedicine.net/app on your smart phone, tablet, or computer. Accreditation: EB Medicine is accredited by the Accreditation Council for Continuing Medical Education (ACCME) to provide continuing medical education for physicians. This activity has been planned and implemented in accordance with the Essential Areas and Policies of the ACCME. Credit Designation: EB Medicine designates this enduring material for a maximum of 4 AMA PRA Category 1 CreditsTM. Physicians should claim only the credit commensurate with the extent of their participation in the activity. ACEP Accreditation: Pediatric Emergency Medicine Practice is also approved by the American College of Emergency Physicians for 48 hours of ACEP Category I credit per annual subscription. AAP Accreditation: This continuing medical education activity has been reviewed by the American Academy of Pediatrics and is acceptable for a maximum of 48 AAP credits per year. These credits can be applied toward the AAP CME/CPD Award available to Fellows and Candidate Fellows of the American Academy of Pediatrics. AOA Accreditation: Pediatric Emergency Medicine Practice is eligible for up to 48 American Osteopathic Association Category 2A or 2B credit hours per year. Needs Assessment: The need for this educational activity was determined by a survey of medical staff, including the editorial board of this publication; review of morbidity and mortality data from the CDC, AHA, NCHS, and ACEP; and evaluation of prior activities for emergency physicians. Target Audience: This enduring material is designed for emergency medicine physicians, physician assistants, nurse practitioners, and residents. Goals: Upon completion of this activity, you should be able to: (1) demonstrate medical decision-making based on the strongest clinical evidence; (2) cost-effectively diagnose and treat the most critical ED presentations; and (3) describe the most common medicolegal pitfalls for each topic covered. Discussion of Investigational Information: As part of the journal, faculty may be presenting investigational information about pharmaceutical products that is outside Food and Drug Administration approved labeling. Information presented as part of this activity is intended solely as continuing medical education and is not intended to promote off-label use of any pharmaceutical product. Faculty Disclosure: It is the policy of EB Medicine to ensure objectivity, balance, independence, transparency, and scientific rigor in all CME-sponsored educational activities. All faculty participating in the planning or implementation of a sponsored activity are expected to disclose to the audience any relevant financial relationships and to assist in resolving any conflict of interest that may arise from the relationship. Presenters must also make a meaningful disclosure to the audience of their discussions of unlabeled or unapproved drugs or devices. In compliance with all ACCME Essentials, Standards, and Guidelines, all faculty for this CME activity were asked to complete a full disclosure statement. The information received is as follows: Dr. Schwartz, Dr. Rubinstein, Dr. Sadowitz, Dr. Werner, Dr. Vella, Dr. Wang, Dr. Damilini, and their related parties report no significant financial interest or other relationship with the manufacturer(s) of any commercial product(s) discussed in this educational presentation. Commercial Support: This issue of Pediatric Emergency Medicine Practice did not receive any commercial support. Earning Credit: Two Convenient Methods: (1) Go online to www.ebmedicine.net/CME and click on the title of this article. (2) Mail or fax the CME Answer And Evaluation Form with your June and December issues to Pediatric Emergency Medicine Practice. Hardware/Software Requirements: You will need a Macintosh or PC with internet capabilities to access the website. Additional Policies: For additional policies, including our statement of conflict of interest, source of funding, statement of informed consent, and statement of human and animal rights, visit http://www.ebmedicine.net/policies. CEO & Publisher: Stephanie Williford Senior Business Analyst: Robin Wilkinson Director of Editorial: Dorothy Whisenhunt Senior Content Editor & CME Director: Erica Scott Content Editor: Cheryl Belton Editorial Content Coordinator: Angie Wallace Director of Operations: Robin Salet Office Manager: Kiana Collier Client Data Associate: Paige Banks Director of Business Development: Susan Woodard Product Marketing & Communications Manager: Kelly Smith Online Marketing Manager: Marcus Snow Client Services Associate: Cory Shrider Direct all inquiries to: EB Medicine Phone: 1-800-249-5770 or 678-366-7933 Fax: 1-770-500-1316 5550 Triangle Parkway, Suite 150 Norcross, GA 30092 E-mail: [email protected] Website: ebmedicine.net To write a letter to the editor, please email: [email protected] Subscription Information Full annual subscription: $319 (includes 12 monthly evidence-based print issues; 48 AMA PRA Category 1 CreditsTM, 48 ACEP Category I credits, 48 AAP Prescribed credits, and 48 AOA Category 2A or 2B CME credits; and full online access to searchable archives and additional CME). Call 1-800- 249-5770 or go to www.ebmedicine.net/subscribe to subscribe. Individual issues: $39 (includes 4 CME credits). Call 1-800-249-5770 or go to www.ebmedicine.net/PEMPissues to order. Group subscriptions at heavily discounted rates are also available. Contact Cory Shrider, Account Manager, at 678-366-7933 x 316 or [email protected] for more information. Pediatric Emergency Medicine Practice (ISSN Print: 1549-9650, ISSN Online: 1549-9669, ACID-FREE) is published monthly (12 times per year) by EB Medicine. 5550 Triangle Parkway, Suite 150, Norcross, GA 30092. Opinions expressed are not necessarily those of this publication. Mention of products or services does not constitute endorsement. This publication is intended as a general guide and is intended to supplement, rather than substitute, professional judgment. It covers a highly technical and complex subject and should not be used for making specific medical decisions. The materials contained herein are not intended to establish policy, procedure, or standard of care. Pediatric Emergency Medicine Practice is a trademark of EB Medicine. Copyright © 2015 EB Medicine All rights reserved. No part of this publication may be reproduced in any format without written consent of EB Medicine. This publication is intended for the use of the individual subscriber only, and may not be copied in whole or in part or redistributed in any way without the publisher’s prior written permission – including reproduction for educational purposes or for internal distribution within a hospital, library, group practice, or other entity. Copyright © 2015 EB Medicine. All rights reserved. 24 Reprints: www.ebmedicine.net/pempissues