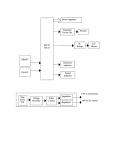
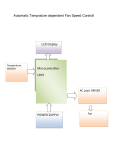
Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Commentary ARTICLE SERIES: Cell Biology and Disease 2319 Microtubule-based transport – basic mechanisms, traffic rules and role in neurological pathogenesis Mariella A. M. Franker1 and Casper C. Hoogenraad1,2,* 1 Cell Biology, Faculty of Science, Utrecht University, 3584 CH Utrecht, The Netherlands Department of Neuroscience, Erasmus Medical Center, 3000 DR Rotterdam, The Netherlands 2 *Author for correspondence ([email protected]) Journal of Cell Science 126, 2319–2329 ß 2013. Published by The Company of Biologists Ltd doi: 10.1242/jcs.115030 Journal of Cell Science Summary Microtubule-based transport is essential for neuronal function because of the large distances that must be traveled by various building blocks and cellular materials. Recent studies in various model systems have unraveled several regulatory mechanisms and traffic rules that control the specificity, directionality and delivery of neuronal cargos. Local microtubule cues, opposing motor activity and cargoadaptors that regulate motor activity control microtubule-based transport in neurons. Impairment of intracellular transport is detrimental to neurons and has emerged as a common factor in several neurological disorders. Genetic approaches have revealed strong links between intracellular transport processes and the pathogenesis of neurological diseases in both the central and peripheral nervous system. This Commentary highlights recent advances in these areas and discusses the transport defects that are associated with the development of neurological diseases. Key words: Brain, Neuron, Neurological disease, Microtubule, Transport, Kinesin, Dynein, Adaptor proteins Introduction To build and maintain highly complex neurons with active synapses, many basic building blocks – for example, proteins, lipids and cellular materials, such as mitochondria and synaptic vesicles – need to be sorted to either axons or dendrites (Kapitein and Hoogenraad, 2011; Namba et al., 2011; Rolls, 2011). It is well known that intracellular cargo transport in neurons is driven by motor proteins that can move directionally along either of two types of cytoskeletal structures: actin filaments and microtubules (see Box 1). Actin facilitates motility of motor proteins of the myosin family, whereas microtubules serve as tracks for two families of motor proteins, the kinesins and dyneins, which move towards the microtubule plus-end or minus-end, respectively (Hirokawa et al., 2009; Schliwa and Woehlke, 2003; Vale, 2003). Evidence shows that microtubule-based transport mainly facilitates the long-range transport into distal axons and dendrites, whereas actin-based transport is important for shortrange transport and delivery of proteins to synapses and growth cones (Dent et al., 2011; Hoogenraad and Bradke, 2009; Kneussel and Wagner, 2013). Although many members of the motor families participate in neuronal cargo trafficking, the diversity of motor proteins alone is insufficient to account for the specificity, directionality and timing of delivery. It has been shown that regulation of transport occurs through several additional processes, such as activation or deactivation of motors, selective binding of motors to cargo, through microtubule-associated proteins (MAPs) and microtubule post-translational modifications (PTMs) (Akhmanova and Hammer, 2010; Goldstein et al., 2008; Janke and Bulinski, 2011; Schlager and Hoogenraad, 2009). How the microtubule-based transport of specific cargo types is spatially and temporally regulated in neurons is now an emerging field of investigation. Neurons are especially vulnerable to defects in transport due to the extreme length of the neuronal processes. The longest axon in the human body can be up to 1 meter long, whereas an average neuronal cell body is ,50 mm. Motor proteins and their regulators have crucial roles in the development of many neurological and neurodegenerative diseases (De Vos et al., 2008; Millecamps and Julien, 2013). Disruption of intracellular transport can result in vesicle-trafficking impairments, alter specific cargo interactions and cause defects in retrograde survival signals, ultimately leading to neuronal death and loss of brain function (Coleman, 2011; Perlson et al., 2010; Saxena and Caroni, 2007). Axonal pathologies, including abnormal accumulations of proteins and organelles have been observed in patients with Huntington’s, Parkinson’s and Alzheimer’s disease, and can severely hinder neuronal transport pathways (Gunawardena and Goldstein, 2005; Millecamps and Julien, 2013). However, whether the accumulations are the cause or the consequence of transport defects is unclear, and the causal relationship between transport impairments and neurodegeneration is unknown. The identification of mutations in genes involved in neuronal transport pathways strongly supports the view that defective transport can directly trigger neurological diseases. Several disruptive mutations in a- or b-tubulin and microtubule-based motors have been directly linked to neurological diseases in both the central and peripheral nervous system (Eschbach and Dupuis, 2011; Millecamps and Julien, 2013; Tischfield et al., 2011). In this Commentary, we will discuss the disease-causing mutations in motor and regulatory proteins that lead to developmental and neurological disorders. Before doing so, we outline the basic molecular mechanisms that control neuronal transport and focus on microtubule cues, bidirectional transport mechanisms and motor adaptors as regulators of motor activity. 2320 Journal of Cell Science 126 (11) Journal of Cell Science Box 1. Biochemical and biophysical properties of motor proteins The movements of organelles were first observed in the squid giant axon and, soon after, molecular motors were identified as the effectors of this transport (Allen et al., 1982; Brady et al., 1982; Vale et al., 1985). The decades following the discovery of motor proteins have yielded many important insights into the molecular, structural and biophysical properties of cytoskeleton-dependent motor transport. Many motor properties, such as directionality, stall forces, and step sizes are now well understood based on controlled in vitro experiments (Block, 2007; Reck-Peterson et al., 2006; Yildiz et al., 2008). The structure of kinesin was first demonstrated using electron microscopy (Hirokawa et al., 2009), and further high-resolution crystallographic structures of the motor domains have led to detailed insights into how force and movement are coupled (Carter et al., 2011; Kon et al., 2012; Kull et al., 1996). Revolutionary single-molecule biophysics experiments established that kinesin walks with an asymmetric hand-over-hand stepping mechanism, hydrolyzing a single ATP molecule per motor step (Asbury et al., 2003; Vale et al., 1996; Yildiz et al., 2004). For the motor to be processive, there must be a proper coordination between the two motor heads. It is still a matter of debate how this coordination, or gating, takes place (Block, 2007). Motor proteins move unidirectionally with either plus-end motility (kinesins) or minus-end motility (dyneins). However, occasional backstepping of motor proteins can occur and was first observed for dynein (Toba et al., 2006). It has been postulated that increased flexibility of the dynein structure gives rise to a diffusional component in the dynein step, causing an ‘irregular’ stepping pattern that allows variable step sizes, back steps and even sideways steps (DeWitt et al., 2012; Qiu et al., 2012). Other studies show that kinesins are also capable of ATPdependent backstepping when they are biased by load (Carter and Cross, 2005). In these situations, the neck-linker region plays an important role in mediating strain between the two motor heads and in regulating gating (Clancy et al., 2011). Transport mechanisms in neurons Microtubule-based transport is crucial for neuronal survival and function, both during development and in the adult brain. Different cellular processes, such as neuronal migration, axon guidance, the establishment and maintenance of neuronal polarity and synaptic plasticity, involve many cytoskeletal elements and transport events that together form the complex cellular networks of the nervous system and ensure that the proper connections are made in the brain (Barnes and Polleux, 2009; Conde and Cáceres, 2009; Hoogenraad and Bradke, 2009). In this section, we will briefly highlight the importance of the cytoskeletal network and motor protein transport for neuronal development and function (Fig. 1). During development of the nervous system, neuron progenitor cells migrate across the brain to their final destination (Hatten, 1999). Although extracellular factors and adhesion molecules provide directional cues to direct the migrating cells and guide axonal growth, cytoskeletal interactions produce the forces that are necessary for nuclear positioning and neuronal migration (Gundersen and Worman, 2013; Kuijpers and Hoogenraad, 2011; Stiess and Bradke, 2011). For example, it has been shown that actin-based myosin motors and the dynein regulator Lis1 (also known as PAFAH1B1) are important for pulling the centrosome and soma forwards during migration (Vallee et al., 2009). Most neurons polarize during the migration process to form a distinct axonal and somatodendritic compartment. It is well known that microtubule-based mechanisms have an active role in determining neuronal polarity and axonal outgrowth (Hoogenraad and Bradke, 2009). For example, stable microtubules can act as specific transport roads for the trafficking of signaling factors that participate in neuronal polarization (Arimura and Kaibuchi, 2007; Witte and Bradke, 2008). After the polarization and differentiation stages, neurons need to maintain their extensively polarized morphology, and specific components must be delivered to the correct neuronal compartment. Specific targeting of different motor–cargo complexes to either the axon or the dendrites ensures that the cargo is transported correctly (Hirokawa et al., 2009; Kapitein and Hoogenraad, 2011). Recent studies have found that, although a few kinesins are nonselective, most kinesin motors preferentially target cargos to the axon (Huang and Banker, 2012) and only dynein transports cargo exclusively into dendrites (Kapitein et al., 2010a). The cytoskeleton organization in the axon initial segment (AIS) is also thought to have a role in axonal targeting by preventing vesicles with dendritic cargo from entering the axon (Song et al., 2009; Watanabe et al., 2012). Moreover, mature neurons form dendritic spines, which are small ‘knob’-shaped extensions protruding from the surface of the dendrites. Dendritic spines are the major post-synaptic sites for excitatory synaptic transmission (Bourne and Harris, 2008; Kennedy et al., 2005). Several studies have shown that dynamic microtubules occasionally enter denditic spines and have a role in spine morphology and synaptic plasticity (Hoogenraad and Akhmanova, 2010). Microtubules in spines might influence actin dynamics and promote the transport of postsynaptic cargos, such as receptors and postsynaptic proteins. Moreover, transport processes in the axon of neurons in the central and peripheral nervous system are of key importance to distribute the various building blocks and cellular materials (Hirokawa et al., 2010; Millecamps and Julien, 2013; Perlson et al., 2010; Saxton and Hollenbeck, 2012). Thus, many different microtubule-based processes are actively involved in various phases of neuronal development and in the mature nervous system. Microtubule cues steer neuronal transport Neuronal transport can be directly regulated by the underlying microtubule cytoskeleton. The local organization of microtubules, MAPs, such as axonal tau, and PTMs all affect for neuronal transport (Janke and Kneussel, 2010; Verhey and Hammond, 2009). In axons, anterograde and retrograde transport is directly dictated by the unipolar organization of the microtubule array (Hirokawa et al., 2009; Hoogenraad and Bradke, 2009). However, in dendrites, the microtubules coalesce into bundles of mixed polarity and a single motor type could mediate bidirectional cargo transport by switching from microtubules in which the minus-end is directed outwards (minus-end out) to microtubules in which the plus-end is directed outwards (plus-end out). The anti-parallel microtubule organization in dendrites provides a structural framework for the regulation of cargo trafficking and raises interesting models with regard to neuronal trafficking rules (Kapitein and Hoogenraad, 2011; Rolls, 2011). Moreover, the influence of the local organization of actin cytoskeleton at presynaptic terminals and dendritic spines and its affect on neuronal cargo transport is of significant importance (Hotulainen and Hoogenraad, 2010; Transport and disease A B Neuronal migration Neuronal polarization Synapse plasticity Mixed microtubules direct dendritic transport Dynein and Lis1 produce pulling force + ++ Dynein C PSD Postsynaptic spine with actin network Actin maintains spine shape and scaffold for the PSD Dendrites – –– – Centrosome – – + + ++ Neuron protrusion – + + Microtubules + – + Nucleus + AIS filters unwanted cargo Actin network Postmitotic neuron Actin and myosin II produce contractile force PSD + PTMs guide axonal transport Axon Microtubules grow inside spines – Radial glial cell Journal of Cell Science 2321 – – – + + Microtubules Microtubule entry influences synaptic activity + – + Key Translocation of nucleus Direction of migration Fig. 1. The role of the cytoskeleton during neuronal development and maturation. (A) During maturation, neurons migrate to their final destination in the brain, establish a highly polarized structure and form chemical synapses that make the connections between the pre- and post-synaptic sites. The cytoskeleton plays crucial roles throughout these processes. During migration, actin-based myosin II motors and microtubule-based dynein motors generate the pulling and pushing forces that translocate the nucleus (see also, Vallee et al., 2009). (B) To maintain the highly polarized neuronal structure, different cargos must be selectively transported to either the somatodendritic or axonal compartment. The panel shows a few important mechanisms that guide neuronal polarity (see also, Kapitein and Hoogenraad, 2011). Microtubule post-translational modification (PTMs) that are present in the axon (blue) but absent in dendrites (red) specifically target axonal cargo, whereas microtubule bundles with mixed orientation direct transport in the dendrites. In addition, the axon initial segment (AIS) filters out dendritic cargo and prevents it from entering the axon. (C) The actin network is the primary component that gives shape to the dendritic spines and forms a scaffold for the post-synaptic density (PSD). In addition, microtubules have been shown to transiently enter spines (see Hoogenraad and Akhmanova, 2010). Kneussel and Wagner, 2013). For example, myosin motors have been shown to counteract microtubule-based transport and facilitate the docking of mitochondria to actin filaments (Saxton and Hollenbeck, 2012). In vitro approaches have shown that the axonal MAP tau can be used to tune the relative amounts of plus-end- and minus-end-directed kinesin transport (Dixit et al., 2008; Vershinin et al., 2007). Low concentrations of tau facilitate kinesin-mediated transport, whereas high concentrations of tau, as is found in the distal parts of the axon, are favorable for dynein-mediated transport. Such a difference between the effects of tau on kinesin- and dynein-based motility has not been confirmed in vivo, where neither overexpression of tau nor tau deletion alters axonal transport (Morfini et al., 2007; Yuan et al., 2008). Interestingly, phosphorylated tau is involved in a number of neurological disorders, such as Alzheimer’s disease, and has recently been shown to mislocalize to dendritic spines and mediate synaptic dysfunction (Hoover et al., 2010). A leading model for driving selective neuronal transport is that the affinity of motor proteins for microtubules is affected by the PTMs on the microtubules (Janke and Kneussel, 2010; Verhey and Hammond, 2009). For example, during neuronal polarization, it is well known that vectorial membrane trafficking occurs into the future axon (Bradke and Dotti, 1997), that stable microtubules act as specific transport roads (Arimura and Kaibuchi, 2007; Witte and Bradke, 2008), and that kinesin-1 motors accumulate specifically at the tips of axons (Jacobson et al., 2006; Nakata and Hirokawa, 2003), all suggesting that kinesin-1 prefers stable microtubules for axonal cargo transport. Recent work has identified a specific region in kinesin-1 that is responsible for its preference to bind to detyrosinated microtubules (Konishi and Setou, 2009). Here, knockdown of tubulin–tyrosin ligase (TTL) to decrease tyrosinated microtubules resulted in the redistribution of kinesin-1 in both axons and dendrites. Other data suggest that the abundance of GTP-tubulin in microtubules at the AIS underlies the selective kinesin-1 localization in axons (Nakata et al., 2011). Therefore, selective transport is probably not regulated by a single factor but probably by various local cues, such as a combination of specific microtubule modifications, associated proteins and tubulin conformations. Opposing motor activity and bidirectional transport Many and diverse types of proteins and cellular materials have been shown to move bidirectionally in cells (Welte, 2004). A large number of organelles and cytoskeleton proteins, including endosomes (Deinhardt et al., 2006), autophagosomes (Maday et al., 2012), intermediate filaments (Li et al., 2012) and synaptic vesicles (Sabo and McAllister, 2003), are known to show bidirectional movement in neuronal axons. Although there are some differences between the movements of various neuronal cargos, they share common features, such as frequent stopping and pausing, as well as reversals in direction during transport, and we are beginning to understand the importance of these mechanisms to achieve appropriate cargo distributions in neurons. An interesting example from Drosophila shows that neuropeptide-containing dense core vesicles, which are transported into the distal axons, initially pass the proximal boutons, the presynaptic axonal bulges. Excess vesicles circulate back to the soma and are occasionally captured in en passant boutons, ensuring uniform distribution throughout the axon (Wong et al., 2012). Although bidirectional movement has been observed in almost all cellular systems, it remains a matter of debate how these processes are regulated. In vitro, motor proteins are intrinsically capable of processively transporting cargo without requiring additional regulation by other factors. As indicate above, a single type of motor protein can mediate bidirectional movement on a mixed network of opposite polarity microtubule bundles, such as that found in the dendrites (Kapitein et al., 2010a). Another interesting aspect of bidirectional transport is the interplay between different microtubule motor types. Backwards and forwards movements are typically observed during neuronal transport; however, net movement in one direction occurs over large distances in neurons. How is this achieved? One possibility is that motors with opposite directionality are continuously acting on a cargo and engage in a stochastic ‘tug-of-war’. This can result in either stalling of the cargo or in bidirectional, salutatory movements depending on the relative ‘strength’ of the attached motors. Ultimately, one type of motor wins and transports the cargo in its preferred direction. A recent study using synthetic ‘DNA origami’ found that ensembles of artificial cargo that are composed of opposite polarity motors are engaged in a tug-of-war, resulting in stalled cargo (Derr et al., 2012). Modeling has shown that stochastic tug-of-war can induce salutatory bidirectional movements (Müller et al., 2008), and deformations of endosomes during transport in vivo in cohort with decreased speeds during transitions between anterograde and retrograde transport (Soppina et al., 2009) argue in favor of this model. In addition, purified neuronal vesicles that move along microtubules in vitro show very similar behavior as observed in vivo, suggesting that all components that are necessary for transport are strongly attached to the vesicles themselves (Hendricks et al., 2010). It is plausible, however, that motoradaptor proteins or other signaling proteins that are attached to the vesicles were co-purified in this study and further regulated the activity of the motors (Gross, 2003; Welte, 2004). Thus, although there is evidence that supports a stochastic ‘tug-of-war’ hypothesis, the activity of opposing motors appears to be closely interlinked (Fig. 2). Knockdown of either kinesin or dynein results in impaired transport in both plus- and minus-end directions in several different cell types (Jolly and Gelfand, 2011), and it has been shown that coupling an active plus-enddirected motor to peroxisomes also induces motility in the opposite direction in Drosophila S2 cells (Ally et al., 2009) suggesting that motors with opposite directionality directly activate each other. Similar results were found in a study on bidirectional movement of mitochondria in Drosophila larval Kinases and phosphatases Rab GTPase Ca2+ Cargo Adaptor protein Dynein – Journal of Cell Science Journal of Cell Science 126 (11) MAPs Microtubule Kinesin PTMs + 2322 Fig. 2. Bidirectional transport. Plus-end-directed kinesin motors and minus-end-directed dynein motors are attached to the same cargo simultaneously and can engage in a stochastic ‘tug-of-war’. In addition, these motors are regulated further to ensure a proper distribution of cargo in living cells (black arrows). Adaptor proteins mediate the specific binding between motor proteins and their cargo. Crosstalk between adaptors and other regulators is also possible, biasing movement in the direction of either kinesin- (blue arrow) or dynein-mediated movement (green arrow). Several signaling events, such as local changes in Ca2+ levels, phosphorylationdependent signaling and regulated Rab GTPase activity can activate or deactivate motors or act on adaptor proteins and other regulators. Finally, motor activity can be regulated by the underlying microtubule cytoskeleton, including microtubule-associated proteins (MAPs) and microtubule posttranslational modifications (PTMs). motor axons (Pilling et al., 2006). In contrast, several other studies report that cargo transport in one direction is readily achieved by selectively targeting a single type of motor protein, and they demonstrate that the activation by a motor with an opposite directionality is not required. For instance, a robust unidirectional motility was observed when kinesin motors are recruited to peroxisomes, even when dynein function was disrupted (Kapitein et al., 2010b). Furthermore, acute inhibition of either dynein heavy chain (DHC) or kinesin-1 heavy chain (KHC, also known as KIF5B) with antibodies has been found to only impair minus-end- and plus-end-directed motion, respectively (Hendricks et al., 2010; Shubeita et al., 2008). These apparently contradictory findings could reflect the various control mechanisms that exist for motor-based protein transport (Jolly and Gelfand, 2011). As will be discussed below in more detail, coordination of opposing motors in neurons is a complex process that is regulated by several molecular machineries at various cellular levels. Motor adaptors as regulators of motor activity It is becoming increasingly clear that interactions between motors and their adaptors are a crucial step in selective transport (Fig. 2) (Akhmanova and Hammer, 2010; Goldstein et al., 2008; Hirokawa et al., 2010; Schlager and Hoogenraad, 2009). A recent report studying the axonal transport of vesicles containing the prion protein (PrPC) showed that the composition of dynein and kinesin motors is the same in anterograde-directed, retrograde-directed and stationary vesicles (Encalada et al., 2011). These data suggest that – in addition to local motor recruitment mechanisms – a stable population of cargoassociated motors can be modulated by regulatory factors to control anterograde and retrograde transport. Indeed, microtubule-based transport is extensively controlled by internal and external factors, and many regulators of dynein and kinesin motors have been identified. Recent research has Journal of Cell Science Transport and disease shown that adaptor proteins that link motors to their cargo can function as either activators or inhibitors of motor proteins and allow for changes in the direction of transport (Akhmanova and Hammer, 2010; Schlager and Hoogenraad, 2009). The list of molecules that are known to link transport motors to specific neuronal cargos is rapidly expanding. Biochemical and proteomics approaches, as well as high-throughput yeast twohybrid screens, have identified more than 100 proteins that are associated with kinesin-1 in mammals, flies and worms (Gindhart, 2006). Most of these proteins are cargo molecules themselves, but some are adaptor proteins, including scaffolding proteins and Rab GTPases that regulate neuronal transport. For example, Jun N-terminal kinase (JNK)-interacting proteins (JIPs) links kinesin-1 to cargo, such as reelin receptor apolipoprotein E receptor 2 (ApoER2, also known as LRP8) and phosphorylated bamyloid precursor protein (APP), and MAP kinase-activating death domain protein (MADD, also known as DENN or Rab3 GDP/GTP exchange factor) acts as an adaptor between kinesin-3 motors and synaptic vesicles (Hirokawa et al., 2010; Namba et al., 2011; Schlager and Hoogenraad, 2009). Moreover, several signaling events, such as local changes in Ca2+ levels, phosphorylation-dependent signaling and regulated Rab GTPase activity, have been found to influence neuronal transport and regulate the loading and unloading of cargo (Fig. 2) (Namba et al., 2011; Saxton and Hollenbeck, 2012; Schlager and Hoogenraad, 2009). Here, we will give two examples of bidirectional cargo adaptors that control the balance between dynein- and kinesin-based transport and regulate selective transport in neurons. Huntingtin, of which the mutated protein is implicated in Huntingon’s disease, acts as a molecular switch between dynein and kinesin-1 motors (Caviston et al., 2011; Colin et al., 2008). When huntingtin is phosphorylated, kinesin is recruited to vesicles, thereby biasing their movement along the microtubule towards the plus-ends. In the unphosphorylated state, kinesin recruitment to the vesicle is no longer stabilized, and dyneinmediated retrograde motility predominates (Colin et al., 2008). Many other lines of research support the view that kinesin- and dynein-mediated transport is modulated by phosphorylation. For example, ERK1/2 phosphorylation of dynein intermediate chain (DIC) recruits cytoplasmic dynein to signaling endosomes for retrograde transport in axons (Mitchell et al., 2012). Moreover, recent studies in Caenorhabditis elegans have shown that two cyclin-dependent kinases, PCT-1 and CDK-5, directly polarize trafficking of presynaptic components by inhibiting dyneinmediated retrograde transport and setting the balance between anterograde and retrograde motors (Ou et al., 2010). In contrast, in rat sensory neurons, CDK5 phosphorylation of the dyneininteracting protein NDEL1 stimulates the cargo transport capacity of dynein (Pandey and Smith, 2011). Adaptor proteins that interact with the mitochondrial outer surface have been identified, and these are potential candidates to regulate mitochondria transport throughout the neuron (Saxton and Hollenbeck, 2012). Recent findings have demonstrated that two very similar motor-adaptor proteins, TRAK1 and TRAK2 utilize different machineries to transport mitochondria to axons and dendrites (van Spronsen et al., 2013). TRAK1 binds to both kinesin-1 and dynein and directs mitochondria into axons, whereas TRAK2 predominantly interacts with dynein–dynactin and mediates the dendritic targeting of mitochondria. These results are consistent with the polarized microtubule organization 2323 in neurons (Baas et al., 1988). Microtubules oriented with the plus-end outwards drive the TRAK1–dynein–kinesin complex into axons, whereas microtubules with mixed orientations allow transport of the TRAK2–dynein complex into dendrites. Interestingly, these data imply that the opposing motors are closely interlinked in axons but can act independently from one another in dendrites. The functional differences between TRAK1 and TRAK2 are explained by conformational differences; the backfolding of TRAK2 affects its interaction with kinesin-1. It is tempting to speculate that conformational switching of motor proteins (see Box 2) and motor-adaptor proteins (as described above) can coordinate bidirectional transport, polarized trafficking and influence local cargo movements. Box 2. Intramolecular motor–tail communication Communication between the motor domain and tail region is an important aspect of motor protein regulation. Recent evidence suggests that the tail region of dynein, through its subunits or associated proteins, controls its own motor activity. In vitro studies found that the legs at odd angles (Loa) mutation F580Y in the tail of the cytoplasmic dynein heavy chain (Hafezparast et al., 2003) inhibits motor processivity (Ori-McKenney et al., 2010). In addition, dynein-associated protein NudE (NDE1) and Lis1 are involved in the interaction between the tail and motor domains of dynein (OriMcKenney et al., 2010; Zylkiewicz et al., 2011). NudE–Lis1mediated communication appears to be achieved through a significantly different mechanism from the folding mechanism for myosin V and some kinesins, which occurs through direct intramolecular interactions between tail and motor domains (Adio et al., 2006; Siththanandan and Sellers, 2011). Kinesin-1 undergoes a conformational change that brings the motor domain in close contact with its tail region as shown by sedimentation analysis (Hackney et al., 1992). This folded, inactive conformation of kinesin-1 blocks processive motor movement (Cai et al., 2007). The crystal structure of kinesin-1 and subsequent biochemical experiments suggest that steric hindrance is not the cause of inhibition, but that instead the tail region restrains ADP release (Kaan et al., 2011). Whereas binding to cargo was initially proposed to unfold kinesin-1 and activate its motility (Coy et al., 1999), it is now thought that motors can be present in an inhibited state on the cargo surface and might alternate between active and inactive conformations in response to external signals (Blasius et al., 2007). In addition, singlemolecule in vitro studies using quantum dots have shown that when kinesin-1 is in a folded inhibited state it is able to passively diffuse along microtubules (Lu et al., 2009). It is plausible that the ability to diffuse along microtubules is a general feature of molecular motors. Similar mechanisms have been described for the kinesin-5 family member Eg5 (also known as KIF11), which is involved in positioning of the mitotic spindle (Kapitein et al., 2008), for KIF1A, a kinesin-3 family member (Hammond et al., 2009), and for MCAK, a kinesin-13 microtubule-depolymerizing motor (Helenius et al., 2006). The motor properties described in Box 1, such as motor back-stepping, motor-neck–tail communication and diffusion might allow motors to slow down and step around obstacles to resolve ‘road blocks’ or ‘traffic jams’, and to switch to other cytoskeletal tracks without exchanging the motors attached to the cargo. Indeed, in disease models of Alzheimer’s and motor neuron disease, it appears that only a small fraction of cargos are trapped in these ‘traffic jams’, suggesting that motors could possibly bypass these blockages (Pilling et al., 2006; Shemesh et al., 2008). 2324 Journal of Cell Science 126 (11) Journal of Cell Science Microtubule transport and disease As microtubule-based transport is fundamental to the development and function of mature neurons, defects in neuronal transport pathways are likely to cause neuronal diseases (Chevalier-Larsen and Holzbaur, 2006; De Vos et al., 2008; Hirokawa et al., 2010; Salinas et al., 2008). Although many studies suggest that transport pathways have a major role in the onset and progression of various human developmental and neurological disorders, the causal relationship between transport impairments and neurodegeneration is largely unclear (Gunawardena and Goldstein, 2005; Millecamps and Julien, 2013). Several kinesin-knockout mice display specific neurological phenotypes, such as epilepsy and hydrocephalus (Nakajima et al., 2012; Niwa et al., 2012), and kinesin-2-related ciliopathy, causing hydrocephalus and disturbance in dorsoventral patterning of the neural tube (Hirokawa et al., 2012). The identification of mutations in genes involved in neuronal transport pathways strengthens the direct link between transport defects and human neurological diseases (Table 1 and Fig. 3). In this section, we will discuss a few examples of how mutations in different tubulin isotypes and motor proteins might lead to defective neuronal transport and trigger neurodegeneration. One way to impair neuronal transport is to disrupt the underlying microtubule network (Baas et al., 2005; Conde and Cáceres, 2009; Janke and Bulinski, 2011; Lumb et al., 2012; Tischfield et al., 2011). Mutations in a- and b-tubulin-encoding genes have been identified in patients suffering from a wide spectrum of neurological disorders, ranging from microcephaly and seizures to intellectual disabilities and peripheral neuropathy. For instance, mutations in the a-tubulin isotype VIII gene TUBA8 cause polymicrogyria with optic nerve hypoplasia (Abdollahi et al., 2009), and mutations in the b-tubulin isotype II gene TUBB2 result in asymmetrical polymicrogyria (Jaglin et al., 2009). Moreover, in mice, mutations in the tubulin-specific chaperone Tbce lead to a severe deficiency of microtubules in distal axons in progressive motor neuronopathy (Martin et al., 2002; Schaefer et al., 2007), and mutations in the microtubule assembly and severing protein spastin 4 (SPG4) cause hereditary spastic paraplegia in humans (Magariello et al., 2013; Nanetti et al., 2012). In addition, missense mutations in TUBB3, which encodes the neuron-specific b-tubulin isotype III, cause a more Table 1. Mutations in transport proteins underlying neurological disorders Mutant protein Functional defect Disease Reference Vesicular and organelle transport Defects in neuronal migration; late-onset motor neuron degeneration Vesicular and organelle transport Synaptic vesicle transport, mitochondrial transport Axonal transport of K+dependent Na+/Ca2+ exchanger (NCKX2) Impaired axonal transport Lissencephaly; Charcot–Marie– Tooth disease 2 (CMT2); Perry syndrome; Spinal muscular atropy Hereditary spastic paraplegia (SPG10); CMT2 CMT2 Congenital fibrosis of the extraocular muscle (CFEOM) (Braunstein et al., 2010; Harms et al., 2010; Weedon et al., 2011; Willemsen et al., 2012) (Crimella et al., 2012; Füger et al., 2012) (Gentil and Cooper, 2012; Zhao et al., 2001) (Desai et al., 2012; Lee et al., 2012) Impaired axonal transport; axonal swelling; synapse retraction Impaired neuronal migration; reduced axonal transport Motor neuron disease; amytrophic lateral sclerosis (ALS); Perry syndrome Lissencephaly (Newsway et al., 2010; Stockmann et al., 2012; Uribe, 2010) (Mokánszki et al., 2012) a-tubulin isoform Neuronal migration Lissencephaly TUBA8 a-tubulin isoform Polymicrogyria with optic nerve hypoplasia TUBB2B b-tubulin isoform Local cell positioning during cerebral cortical development Neuronal migration (Cushion et al., 2013; Hikita et al., 2013; Tischfield et al., 2011) (Abdollahi et al., 2009; Tischfield et al., 2011) TUBB3 b-tubulin isoform (neuron specific) Neuronal migrations and axon guidance defects TUBB5 TBCE b-tubulin isoform Tubulin chaperone Neuronal migration Impaired maintenance of microtubules in motor axons Synaptic growth and neurotransmission defects CFEOM Facial paralysis Late-onset axonal sensorimotor polyneuropathy Microcephaly Progressive motor neuropathy Mutations in motors DYNC1H1 (dynein) KIF5A KIF1B KIF21A Motor adaptor and regulator protein mutations DCTN1/p150glued (dynactin) LIS1 (PAFAH1B1) Cytoskeletal mutations TUBA1A SPG4 (spastin) Functional role Dynein regulation Coupling of nucleus to centrosome during neuronal migration; dynein regulator Microtubule severer Reduced transport of synaptic vesicle proteins Atrophy of ocular motor muscles Asymmetrical polymicrogyria Hereditary spastic paraplegia (Cushion et al., 2013; Romaniello et al., 2012; Tischfield et al., 2011) (Poirier et al., 2010; Tischfield et al., 2011) (Breuss et al., 2012) (Martin et al., 2002; Schaefer et al., 2007) (Magariello et al., 2013; Nanetti et al., 2012) The table is limited to mutations in motor proteins and associated proteins that have a strong genetic link to neurological disease. Transport and disease 2325 Tubulin α-tubulin β-tubulin TUBB5 TUBB2B TUBB3 Spastin TUBA8 TUBA1A Dendrite – + Microtubule TBCE Microencephaly Polymicrogyria Lissencephaly Congenital fibrosis of the extraocular muscle (CFEOM) Motor neuropathy Hereditary spastic paraplegia (HSP) Axon Dendrite Kinesin Dynein Mitochondrion Amyotrohic lateral sclerosis (ALS) Cargo Charcot–Marie– Tooth 2 (CMT2) Dynein Dynactin Lis1 + – Microtubule KIF1B – + Microtubule Perry syndrome Charcot–Marie– Tooth 2 (CMT2) Hereditary spastic paraplegia (HSP) Congenital fibrosis of the extraocular muscle (CFEOM) Spinal muscular atrophy (SMA) KIF5A KIF21A – Journal of Cell Science Lissencenphaly + Microtubule diffuse spectrum of brain malformations and neurological disabilities, and certain phenotypes often segregate with particular amino acid substitutions (Poirier et al., 2010; Tischfield et al., 2010). A recent study directly investigated whether TUBB3 mutations impair microtubule-based transport in neurons and found that of the 14 b-tubulin mutants tested, only two TUBB3 mutations directly disrupt the interaction with kinesin motors and subsequently impair axonal transport (Niwa et al., 2013). The other mutations in b-tubulin III probably disturb other microtubule properties, such as the stability of a-tubulin–btubulin heterodimers or other characteristics, including the (de)polymerization properties of tubulin, microtubule dynamics or MAP interactions (Tischfield et al., 2011). Several mutations in microtubule-based motors have also been directly linked to neurological diseases. These motor mutations can affect specific neuronal transport routes, or involve multiple cargos and impair multiple pathways (Chevalier-Larsen and Holzbaur, 2006; Hirokawa et al., 2010). Kinesin motor proteins have been directly linked to several neurodegenerative diseases. Mutations in the motor and stalk domain of the kinesin-1 family member KIF5A cause hereditary spastic paraplegia and Charcot– Marie–Tooth (CMT) disease type 2 (Crimella et al., 2012; Reid et al., 2002). Mutations in the KIF21A stalk region have been shown to be the underlying cause of congenital fibrosis of the extraocular muscle type 1 (CFEOM1) (Yamada et al., 2003), which is associated with defects in the oculomotor nerve and results in an inability to move or raise the eyes. A recent study Fig. 3. Major transport defects in neurons and neurological diseases. Impaired neuronal transport can be caused by disruption of the microtubule network, by changes in the processivity or activation of motor proteins, or by effects on the binding of cargo to motor proteins. As illustrated here, interfering with either of these regulatory mechanisms gives rise to neuronal transport defects and often leads to variety of neurological disease phenotypes. For example, mutations in the kinesin motor protein KIF5A have been linked to dominant forms of the hereditary spastic paraplegia (HSP) and Charcot–Marie–Tooth disease type 2 (CMT2) in humans, whereas mutations in b-tubulin isotype III gene TUBB3 leads to the ocular motility disorder, congenital fibrosis of the extraocular muscles type 3 (CFEOM3). Furthermore, mutations in dynein and its accessory proteins dynactin and Lis1 have been associated to spinal muscular atrophy (SMA), amytrophic lateral sclerosis (ALS)-like motor neuron degeneration and earlyonset parkinsonism called Perry syndrome. showed that knockdown of KIF21A causes the dysregulation of Ca2+ at axonal boutons owing to inhibited transport of NCKX2, a K+-dependent Na+-exchanger that is involved in Ca2+ clearance in the large axon terminals of central neurons (Lee et al., 2012). Moreover, a mutation in KIF1B-b, a kinesin-3 family member, was identified in CMT2 patients (Zhao et al., 2001). The heterogyzous Kif1b-knockout mouse presents similar phenotypes, including a chronic peripheral neuropathy with progressive motor weakness and motor coordination, and investigation of its sciatic nerves revealed that transport of synaptic vesicles to the distal axon is impaired. The KIF1B-b mutation is not the only gene that is linked to CMT2, because, so far, mutations in more than 30 genes have been identified that cause CMT disease, several of which are linked to specific transport pathways (Gentil and Cooper, 2012). Cytoplasmic dynein has a pivotal role in neuronal trafficking because it is the only microtubule-based motor protein with minus-end-directed motility (Pfister et al., 2006). Indeed, mutations in the dynein heavy chain gene DHC1H1 lead to striatal atrophy, neuronal migration defects, intellectual disability, CMT and spinal muscular atrophy (SMA) (Braunstein et al., 2010; Harms et al., 2010; Weedon et al., 2011; Willemsen et al., 2012). Consistent with these findings, mutations in dynein in Drosophila and C. elegans result in intracellular protein aggregates and behavioral abnormalities, including locomotor impairment (Eaton et al., 2002; Koushika et al., 2004). Furthermore, disrupting the function of dynein in Journal of Cell Science 2326 Journal of Cell Science 126 (11) motor neurons in mice causes amyotrophic lateral sclerosis (ALS)-like features (LaMonte et al., 2002; Teuling et al., 2008). Mice strains with spontaneous heterozygous mutations in DYNC1H1, such as the ‘legs at odd angles’ (Loa) and ‘cramping 1’ (Cra1) alleles, also show late-onset motor neuron degeneration (Hafezparast et al., 2003. Recent in vitro studies found that the Loa mutation F580Y in the tail of the mouse cytoplasmic dynein heavy chain inhibits motor processivity (OriMcKenney et al., 2010). These data indicate that communication between the motor domain and tail region is another important aspect of motor regulation (see Box 2). Dynein is regulated by a number of factors, such as LIS1, which, when disrupted, have also been implicated in neurodevelopmental and neurological diseases (Kardon and Vale, 2009). For example, missense mutations in the dynactin gene (DCTN1) that encodes the p150glued subunit of dynactin have been associated with both familial and sporadic ALS (Münch et al., 2004; Puls et al., 2003; Puls et al., 2005). Neuronal expression of mutant p150glued in mice causes motor neuron disease, which is characterized by defects in vesicular transport, axonal swelling and axo-terminal degeneration (Chevalier-Larsen and Holzbaur, 2006; Lai et al., 2007; Laird et al., 2008). It has recently been hypothesized that the disease-causing mutations in the CAP-Gly domain of p150glued inhibit the binding of dynactin at microtubule plusends, thereby abolishing the initiation of retrograde axonal transport (Lloyd et al., 2012; Moughamian and Holzbaur, 2012). Interestingly, different mutations in the same microtubulebinding domain of p150glued cause Perry syndrome, a Parkinson-like disease that is characterized by the loss of dopaminergic neurons (Farrer et al., 2009). Another well-known neurological disorder that has been linked to the dynein motor complex is lissencephaly, a developmental brain malformation in which the surface of the brain is smooth rather than full of folds and the disease subsequently leads to seizures and mental retardation (Kato and Dobyns, 2003; Kuijpers and Hoogenraad, 2011; Métin et al., 2008; WynshawBoris et al., 2010). Perturbing the levels of Lis1 causes defects in microtubule-based processes, such as nuclear migration and cargo transport (Pandey and Smith, 2011; Tanaka et al., 2004; Tsai et al., 2007). Lis1 has been found to function in microtubulebased trafficking by preparing dynein-coupled cargos for transport by targeting it to the plus-ends of microtubules (Egan et al., 2012; Li et al., 2005; McKenney et al., 2010; Yi et al., 2011). Recent single-molecule and structural studies demonstrated that Lis1 binds in between the ATPase and microtubule-binding domains of dynein and prolongs its attachments to microtubules (Huang and Banker, 2012). Lis1 – at least in yeast – is not required for dynein-based cargo motility once dynein is moving, but it instead has a general role in initiating dynein-driven motility (Egan et al., 2012). The numerous mutations in dynein, dynactin and associated proteins, and the broad spectrum of phenotypes, demand a clinical re-classification of dynein-related disorders, where the neurological phenotype must be defined not only genetically but also by the specific cellular effects. Future studies should aim at dissecting the complexities of the dynein structure and its interaction with associated proteins to find out the various mechanisms by which disease-related dynein mutations disrupt transport functions in the central and peripheral nervous system. Conclusions Transport is essential for many different processes in neurons in order to establish and maintain polarity, for axon guidance and regeneration, as well as for synaptic plasticity, both during developmental stages and in the adult brain. Although the biochemical mechanisms underlying motor proteins-based transport are well understood, much of the regulatory processes that act on these motors are still unknown. Many studies now show the importance of these regulatory pathways and emphasize the role of adaptor proteins and regulators, in particular with relevance to diseases of the brain. However, a number of questions remain. How does a motor find the appropriate cargo? How many different motor types are attached to a specific cargo? How does the cargo know when to stop and ‘unload’ its content? Disruption of transport is thought to be an early and perhaps causative event in Alzheimer’s disease, Huntington’s disease and ALS, in which abnormal accumulations of cytoskeleton proteins and organelles in axons have been observed (De Vos et al., 2008; Millecamps and Julien, 2013). The pivotal challenges are to identify the molecular and cellular cause of human brain disorders that arise from intracellular transport defects and to determine how changes in motor protein regulation impact on neuronal structure and function. Defects in transport lead to many different and stress-related responses in cells, making it difficult to elucidate the exact mechanisms underlying a disease. To better understand intracellular transport in neurons, a better understanding of the organization and dynamics of underlying microtubule cytoskeleton, as well as novel approaches to control and manipulate neuronal cargo transport are required (Jenkins et al., 2012; Kapitein et al., 2010a). The spatial and temporal arrangement of transport complexes and the cytoskeleton along which they move is of crucial importance in order to understand how multiple motors can regulate the transport and delivery of specific cargos. These findings could potentially lead to new tools that will be useful when investigating the transport machinery in neurodegenerative disease models. Future studies using genetic disease mouse models will advance our understanding of the neurodegenerative disorders. Functional assays in primary neuronal cells from mouse models or neurons developed from inducible pluripotent stem (IPS) cells of patients (Abeliovich and Doege, 2009) will further help to elucidate the specific disease mechanisms. Moreover, advances in the discovery and synthesis of small molecules that can modulate microtubule-based processes in the brain might offer new therapeutic paradigms to treat neurological defects and intervene in neurodegenerative processes. The use of the microtubule-stabilizing drug taxol to decrease fibrotic scar formation and promote axon regeneration in rodents after spinal cord injury (Ertürk et al., 2007; Hellal et al., 2011) is a promising step in this direction, and other similar approaches hopefully will follow. Acknowledgements We thank Lukas Kapitein and Phebe Wulf for critical reading of the manuscript. Funding The work of our laboratories is supported by Het Prinses Beatrix Spierfonds; the Netherlands Organization for Scientific Research (NWO-ALW-VICI); the Netherlands Organization for Health Research and Development (ZonMW-TOP); the European Science Transport and disease Foundation (EURYI); and the EMBO Young Investigators Program (YIP). Journal of Cell Science References Abdollahi, M. R., Morrison, E., Sirey, T., Molnar, Z., Hayward, B. E., Carr, I. M., Springell, K., Woods, C. G., Ahmed, M., Hattingh, L. et al. (2009). Mutation of the variant alpha-tubulin TUBA8 results in polymicrogyria with optic nerve hypoplasia. Am. J. Hum. Genet. 85, 737-744. Abeliovich, A. and Doege, C. A. (2009). Reprogramming therapeutics: iPS cell prospects for neurodegenerative disease. Neuron 61, 337-339. Adio, S., Reth, J., Bathe, F. and Woehlke, G. (2006). Review: regulation mechanisms of Kinesin-1. J. Muscle Res. Cell Motil. 27, 153-160. Akhmanova, A. and Hammer, J. A., III (2010). Linking molecular motors to membrane cargo. Curr. Opin. Cell Biol. 22, 479-487. Allen, R. D., Metuzals, J., Tasaki, I., Brady, S. T. and Gilbert, S. P. (1982). Fast axonal transport in squid giant axon. Science 218, 1127-1129. Ally, S., Larson, A. G., Barlan, K., Rice, S. E. and Gelfand, V. I. (2009). Oppositepolarity motors activate one another to trigger cargo transport in live cells. J. Cell Biol. 187, 1071-1082. Arimura, N. and Kaibuchi, K. (2007). Neuronal polarity: from extracellular signals to intracellular mechanisms. Nat. Rev. Neurosci. 8, 194-205. Asbury, C. L., Fehr, A. N. and Block, S. M. (2003). Kinesin moves by an asymmetric hand-over-hand mechanism. Science 302, 2130-2134. Baas, P. W., Deitch, J. S., Black, M. M. and Banker, G. A. (1988). Polarity orientation of microtubules in hippocampal neurons: uniformity in the axon and nonuniformity in the dendrite. Proc. Natl. Acad. Sci. USA 85, 8335-8339. Baas, P. W., Karabay, A. and Qiang, L. (2005). Microtubules cut and run. Trends Cell Biol. 15, 518-524. Barnes, A. P. and Polleux, F. (2009). Establishment of axon-dendrite polarity in developing neurons. Annu. Rev. Neurosci. 32, 347-381. Blasius, T. L., Cai, D., Jih, G. T., Toret, C. P. and Verhey, K. J. (2007). Two binding partners cooperate to activate the molecular motor Kinesin-1. J. Cell Biol. 176, 11-17. Block, S. M. (2007). Kinesin motor mechanics: binding, stepping, tracking, gating, and limping. Biophys. J. 92, 2986-2995. Bourne, J. N. and Harris, K. M. (2008). Balancing structure and function at hippocampal dendritic spines. Annu. Rev. Neurosci. 31, 47-67. Bradke, F. and Dotti, C. G. (1997). Neuronal polarity: vectorial cytoplasmic flow precedes axon formation. Neuron 19, 1175-1186. Brady, S. T., Lasek, R. J. and Allen, R. D. (1982). Fast axonal transport in extruded axoplasm from squid giant axon. Science 218, 1129-1131. Braunstein, K. E., Eschbach, J., Ròna-Vörös, K., Soylu, R., Mikrouli, E., Larmet, Y., René, F., Gonzalez De Aguilar, J. L., Loeffler, J. P., Müller, H. P. et al. (2010). A point mutation in the dynein heavy chain gene leads to striatal atrophy and compromises neurite outgrowth of striatal neurons. Hum. Mol. Genet. 19, 4385-4398. Breuss, M., Heng, J. I., Poirier, K., Tian, G., Jaglin, X. H., Qu, Z., Braun, A., Gstrein, T., Ngo, L., Haas, M. et al. (2012). Mutations in the b-tubulin gene TUBB5 cause microcephaly with structural brain abnormalities. Cell Rep. 2, 1554-1562. Cai, D., Hoppe, A. D., Swanson, J. A. and Verhey, K. J. (2007). Kinesin-1 structural organization and conformational changes revealed by FRET stoichiometry in live cells. J. Cell Biol. 176, 51-63. Carter, N. J. and Cross, R. A. (2005). Mechanics of the kinesin step. Nature 435, 308312. Carter, A. P., Cho, C., Jin, L. and Vale, R. D. (2011). Crystal structure of the dynein motor domain. Science 331, 1159-1165. Caviston, J. P., Zajac, A. L., Tokito, M. and Holzbaur, E. L. (2011). Huntingtin coordinates the dynein-mediated dynamic positioning of endosomes and lysosomes. Mol. Biol. Cell 22, 478-492. Chevalier-Larsen, E. and Holzbaur, E. L. (2006). Axonal transport and neurodegenerative disease. Biochim. Biophys. Acta 1762, 1094-1108. Clancy, B. E., Behnke-Parks, W. M., Andreasson, J. O., Rosenfeld, S. S. and Block, S. M. (2011). A universal pathway for kinesin stepping. Nat. Struct. Mol. Biol. 18, 1020-1027. Coleman, M. (2011). Molecular signaling how do axons die? Adv. Genet. 73, 185-217. Colin, E., Zala, D., Liot, G., Rangone, H., Borrell-Pagès, M., Li, X. J., Saudou, F. and Humbert, S. (2008). Huntingtin phosphorylation acts as a molecular switch for anterograde/retrograde transport in neurons. EMBO J. 27, 2124-2134. Conde, C. and Cáceres, A. (2009). Microtubule assembly, organization and dynamics in axons and dendrites. Nat. Rev. Neurosci. 10, 319-332. Coy, D. L., Hancock, W. O., Wagenbach, M. and Howard, J. (1999). Kinesin’s tail domain is an inhibitory regulator of the motor domain. Nat. Cell Biol. 1, 288-292. Crimella, C., Baschirotto, C., Arnoldi, A., Tonelli, A., Tenderini, E., Airoldi, G., Martinuzzi, A., Trabacca, A., Losito, L., Scarlato, M. et al. (2012). Mutations in the motor and stalk domains of KIF5A in spastic paraplegia type 10 and in axonal Charcot-Marie-Tooth type 2. Clin. Genet. 82, 157-164. Cushion, T. D., Dobyns, W. B., Mullins, J. G., Stoodley, N., Chung, S. K., Fry, A. E., Hehr, U., Gunny, R., Aylsworth, A. S., Prabhakar, P. et al. (2013). Overlapping cortical malformations and mutations in TUBB2B and TUBA1A. Brain 136, 536-548. De Vos, K. J., Grierson, A. J., Ackerley, S. and Miller, C. C. (2008). Role of axonal transport in neurodegenerative diseases. Annu. Rev. Neurosci. 31, 151-173. Deinhardt, K., Salinas, S., Verastegui, C., Watson, R., Worth, D., Hanrahan, S., Bucci, C. and Schiavo, G. (2006). Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron 52, 293-305. 2327 Dent, E. W., Gupton, S. L. and Gertler, F. B. (2011). The growth cone cytoskeleton in axon outgrowth and guidance. Cold Spring Harb. Perspect. Biol. 3, a001800. Derr, N. D., Goodman, B. S., Jungmann, R., Leschziner, A. E., Shih, W. M. and Reck-Peterson, S. L. (2012). Tug-of-war in motor protein ensembles revealed with a programmable DNA origami scaffold. Science 338, 662-665. Desai, J., Velo, M. P., Yamada, K., Overman, L. M. and Engle, E. C. (2012). Spatiotemporal expression pattern of KIF21A during normal embryonic development and in congenital fibrosis of the extraocular muscles type 1 (CFEOM1). Gene Expr. Patterns 12, 180-188. DeWitt, M. A., Chang, A. Y., Combs, P. A. and Yildiz, A. (2012). Cytoplasmic dynein moves through uncoordinated stepping of the AAA+ ring domains. Science 335, 221225. Dixit, R., Ross, J. L., Goldman, Y. E. and Holzbaur, E. L. (2008). Differential regulation of dynein and kinesin motor proteins by tau. Science 319, 1086-1089. Eaton, B. A., Fetter, R. D. and Davis, G. W. (2002). Dynactin is necessary for synapse stabilization. Neuron 34, 729-741. Egan, M. J., Tan, K. and Reck-Peterson, S. L. (2012). Lis1 is an initiation factor for dynein-driven organelle transport. J. Cell Biol. 197, 971-982. Encalada, S. E., Szpankowski, L., Xia, C. H. and Goldstein, L. S. (2011). Stable kinesin and dynein assemblies drive the axonal transport of mammalian prion protein vesicles. Cell 144, 551-565. Ertürk, A., Hellal, F., Enes, J. and Bradke, F. (2007). Disorganized microtubules underlie the formation of retraction bulbs and the failure of axonal regeneration. J. Neurosci. 27, 9169-9180. Eschbach, J. and Dupuis, L. (2011). Cytoplasmic dynein in neurodegeneration. Pharmacol. Ther. 130, 348-363. Farrer, M. J., Hulihan, M. M., Kachergus, J. M., Dächsel, J. C., Stoessl, A. J., Grantier, L. L., Calne, S., Calne, D. B., Lechevalier, B., Chapon, F. et al. (2009). DCTN1 mutations in Perry syndrome. Nat. Genet. 41, 163-165. Füger, P., Sreekumar, V., Schüle, R., Kern, J. V., Stanchev, D. T., Schneider, C. D., Karle, K. N., Daub, K. J., Siegert, V. K., Flötenmeyer, M. et al. (2012). Spastic paraplegia mutation N256S in the neuronal microtubule motor KIF5A disrupts axonal transport in a Drosophila HSP model. PLoS Genet. 8, e1003066. Gentil, B. J. and Cooper, L. (2012). Molecular basis of axonal dysfunction and traffic impairments in CMT. Brain Res. Bull. 88, 444-453. Gindhart, J. G. (2006). Towards an understanding of kinesin-1 dependent transport pathways through the study of protein-protein interactions. Brief Funct. Genomic Proteomic 5, 74-86. Goldstein, A. Y., Wang, X. and Schwarz, T. L. (2008). Axonal transport and the delivery of pre-synaptic components. Curr. Opin. Neurobiol. 18, 495-503. Gross, S. P. (2003). Dynactin: coordinating motors with opposite inclinations. Curr. Biol. 13, R320-R322. Gunawardena, S. and Goldstein, L. S. (2005). Polyglutamine diseases and transport problems: deadly traffic jams on neuronal highways. Arch. Neurol. 62, 46-51. Gundersen, G. G. and Worman, H. J. (2013). Nuclear positioning. Cell 152, 13761389. Hackney, D. D., Levitt, J. D. and Suhan, J. (1992). Kinesin undergoes a 9 S to 6 S conformational transition. J. Biol. Chem. 267, 8696-8701. Hafezparast, M., Klocke, R., Ruhrberg, C., Marquardt, A., Ahmad-Annuar, A., Bowen, S., Lalli, G., Witherden, A. S., Hummerich, H., Nicholson, S. et al. (2003). Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science 300, 808-812. Hammond, J. W., Cai, D., Blasius, T. L., Li, Z., Jiang, Y., Jih, G. T., Meyhofer, E. and Verhey, K. J. (2009). Mammalian Kinesin-3 motors are dimeric in vivo and move by processive motility upon release of autoinhibition. PLoS Biol. 7, e72. Harms, M. B., Allred, P., Gardner, R., Jr, Fernandes Filho, J. A., Florence, J., Pestronk, A., Al-Lozi, M. and Baloh, R. H. (2010). Dominant spinal muscular atrophy with lower extremity predominance: linkage to 14q32. Neurology 75, 539546. Hatten, M. E. (1999). Central nervous system neuronal migration. Annu. Rev. Neurosci. 22, 511-539. Helenius, J., Brouhard, G., Kalaidzidis, Y., Diez, S. and Howard, J. (2006). The depolymerizing kinesin MCAK uses lattice diffusion to rapidly target microtubule ends. Nature 441, 115-119. Hellal, F., Hurtado, A., Ruschel, J., Flynn, K. C., Laskowski, C. J., Umlauf, M., Kapitein, L. C., Strikis, D., Lemmon, V., Bixby, J. et al. (2011). Microtubule stabilization reduces scarring and causes axon regeneration after spinal cord injury. Science 331, 928-931. Hendricks, A. G., Perlson, E., Ross, J. L., Schroeder, H. W., III, Tokito, M. and Holzbaur, E. L. (2010). Motor coordination via a tug-of-war mechanism drives bidirectional vesicle transport. Curr. Biol. 20, 697-702. Hikita, N., Hattori, H., Kato, M., Sakuma, S., Morotomi, Y., Ishida, H., Seto, T., Tanaka, K., Shimono, T., Shintaku, H. et al. (2013). A case of TUBA1A mutation presenting with lissencephaly and Hirschsprung disease. Brain Dev. Hirokawa, N., Noda, Y., Tanaka, Y. and Niwa, S. (2009). Kinesin superfamily motor proteins and intracellular transport. Nat. Rev. Mol. Cell Biol. 10, 682-696. Hirokawa, N., Niwa, S. and Tanaka, Y. (2010). Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron 68, 610638. Hirokawa, N., Tanaka, Y. and Okada, Y. (2012). Cilia, KIF3 molecular motor and nodal flow. Curr. Opin. Cell Biol. 24, 31-39. Hoogenraad, C. C. and Akhmanova, A. (2010). Dendritic spine plasticity: new regulatory roles of dynamic microtubules. Neuroscientist 16, 650-661. Journal of Cell Science 2328 Journal of Cell Science 126 (11) Hoogenraad, C. C. and Bradke, F. (2009). Control of neuronal polarity and plasticity—a renaissance for microtubules? Trends Cell Biol. 19, 669-676. Hoover, B. R., Reed, M. N., Su, J., Penrod, R. D., Kotilinek, L. A., Grant, M. K., Pitstick, R., Carlson, G. A., Lanier, L. M., Yuan, L. L. et al. (2010). Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68, 1067-1081. Hotulainen, P. and Hoogenraad, C. C. (2010). Actin in dendritic spines: connecting dynamics to function. J. Cell Biol. 189, 619-629. Huang, C. F. and Banker, G. (2012). The translocation selectivity of the kinesins that mediate neuronal organelle transport. Traffic 13, 549-564. Jacobson, C., Schnapp, B. and Banker, G. A. (2006). A change in the selective translocation of the Kinesin-1 motor domain marks the initial specification of the axon. Neuron 49, 797-804. Jaglin, X. H., Poirier, K., Saillour, Y., Buhler, E., Tian, G., Bahi-Buisson, N., FalletBianco, C., Phan-Dinh-Tuy, F., Kong, X. P., Bomont, P. et al. (2009). Mutations in the beta-tubulin gene TUBB2B result in asymmetrical polymicrogyria. Nat. Genet. 41, 746-752. Janke, C. and Bulinski, J. C. (2011). Post-translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat. Rev. Mol. Cell Biol. 12, 773-786. Janke, C. and Kneussel, M. (2010). Tubulin post-translational modifications: encoding functions on the neuronal microtubule cytoskeleton. Trends Neurosci. 33, 362-372. Jenkins, B., Decker, H., Bentley, M., Luisi, J. and Banker, G. (2012). A novel split kinesin assay identifies motor proteins that interact with distinct vesicle populations. J. Cell Biol. 198, 749-761. Jolly, A. L. and Gelfand, V. I. (2011). Bidirectional intracellular transport: utility and mechanism. Biochem. Soc. Trans. 39, 1126-1130. Kaan, H. Y., Hackney, D. D. and Kozielski, F. (2011). The structure of the kinesin-1 motor-tail complex reveals the mechanism of autoinhibition. Science 333, 883-885. Kapitein, L. C. and Hoogenraad, C. C. (2011). Which way to go? Cytoskeletal organization and polarized transport in neurons. Mol. Cell. Neurosci. 46, 9-20. Kapitein, L. C., Kwok, B. H., Weinger, J. S., Schmidt, C. F., Kapoor, T. M. and Peterman, E. J. (2008). Microtubule cross-linking triggers the directional motility of kinesin-5. J. Cell Biol. 182, 421-428. Kapitein, L. C., Schlager, M. A., Kuijpers, M., Wulf, P. S., van Spronsen, M., MacKintosh, F. C. and Hoogenraad, C. C. (2010a). Mixed microtubules steer dynein-driven cargo transport into dendrites. Curr. Biol. 20, 290-299. Kapitein, L. C., Schlager, M. A., van der Zwan, W. A., Wulf, P. S., Keijzer, N. and Hoogenraad, C. C. (2010b). Probing intracellular motor protein activity using an inducible cargo trafficking assay. Biophys. J. 99, 2143-2152. Kardon, J. R. and Vale, R. D. (2009). Regulators of the cytoplasmic dynein motor. Nat. Rev. Mol. Cell Biol. 10, 854-865. Kato, M. and Dobyns, W. B. (2003). Lissencephaly and the molecular basis of neuronal migration. Hum. Mol. Genet. 12 Spec No 1, R89-R96. Kennedy, M. B., Beale, H. C., Carlisle, H. J. and Washburn, L. R. (2005). Integration of biochemical signalling in spines. Nat. Rev. Neurosci. 6, 423-434. Kneussel, M. and Wagner, W. (2013). Myosin motors at neuronal synapses: drivers of membrane transport and actin dynamics. Nat. Rev. Neurosci. 14, 233-247. Kon, T., Oyama, T., Shimo-Kon, R., Imamula, K., Shima, T., Sutoh, K. and Kurisu, G. (2012). The 2.8 Å crystal structure of the dynein motor domain. Nature 484, 345350. Konishi, Y. and Setou, M. (2009). Tubulin tyrosination navigates the kinesin-1 motor domain to axons. Nat. Neurosci. 12, 559-567. Koushika, S. P., Schaefer, A. M., Vincent, R., Willis, J. H., Bowerman, B. and Nonet, M. L. (2004). Mutations in Caenorhabditis elegans cytoplasmic dynein components reveal specificity of neuronal retrograde cargo. J. Neurosci. 24, 39073916. Kuijpers, M. and Hoogenraad, C. C. (2011). Centrosomes, microtubules and neuronal development. Mol. Cell. Neurosci. 48, 349-358. Kull, F. J., Sablin, E. P., Lau, R., Fletterick, R. J. and Vale, R. D. (1996). Crystal structure of the kinesin motor domain reveals a structural similarity to myosin. Nature 380, 550-555. Lai, C., Lin, X., Chandran, J., Shim, H., Yang, W. J. and Cai, H. (2007). The G59S mutation in p150(glued) causes dysfunction of dynactin in mice. J. Neurosci. 27, 13982-13990. Laird, F. M., Farah, M. H., Ackerley, S., Hoke, A., Maragakis, N., Rothstein, J. D., Griffin, J., Price, D. L., Martin, L. J. and Wong, P. C. (2008). Motor neuron disease occurring in a mutant dynactin mouse model is characterized by defects in vesicular trafficking. J. Neurosci. 28, 1997-2005. LaMonte, B. H., Wallace, K. E., Holloway, B. A., Shelly, S. S., Ascaño, J., Tokito, M., Van Winkle, T., Howland, D. S. and Holzbaur, E. L. (2002). Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron 34, 715-727. Lee, K. H., Lee, J. S., Lee, D., Seog, D. H., Lytton, J., Ho, W. K. and Lee, S. H. (2012). KIF21A-mediated axonal transport and selective endocytosis underlie the polarized targeting of NCKX2. J. Neurosci. 32, 4102-4117. Li, J., Lee, W. L. and Cooper, J. A. (2005). NudEL targets dynein to microtubule ends through LIS1. Nat. Cell Biol. 7, 686-690. Li, Y., Jung, P. and Brown, A. (2012). Axonal transport of neurofilaments: a single population of intermittently moving polymers. J. Neurosci. 32, 746-758. Lloyd, T. E., Machamer, J., O’Hara, K., Kim, J. H., Collins, S. E., Wong, M. Y., Sahin, B., Imlach, W., Yang, Y., Levitan, E. S. et al. (2012). The p150(Glued) CAP-Gly domain regulates initiation of retrograde transport at synaptic termini. Neuron 74, 344-360. Lu, H., Ali, M. Y., Bookwalter, C. S., Warshaw, D. M. and Trybus, K. M. (2009). Diffusive movement of processive kinesin-1 on microtubules. Traffic 10, 1429-1438. Lumb, J. H., Connell, J. W., Allison, R. and Reid, E. (2012). The AAA ATPase spastin links microtubule severing to membrane modelling. Biochim. Biophys. Acta 1823, 192-197. Maday, S., Wallace, K. E. and Holzbaur, E. L. (2012). Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J. Cell Biol. 196, 407-417. Magariello, A., Tortorella, C., Patitucci, A., Tortelli, R., Liguori, M., Mazzei, R., Conforti, F. L., Citrigno, L., Ungaro, C., Simone, I. L. et al. (2013). First mutation in the nuclear localization signal sequence of spastin protein identified in a patient with hereditary spastic paraplegia. Eur. J. Neurol. 20, e22-e23. Martin, N., Jaubert, J., Gounon, P., Salido, E., Haase, G., Szatanik, M. and Guénet, J. L. (2002). A missense mutation in Tbce causes progressive motor neuronopathy in mice. Nat. Genet. 32, 443-447. McKenney, R. J., Vershinin, M., Kunwar, A., Vallee, R. B. and Gross, S. P. (2010). LIS1 and NudE induce a persistent dynein force-producing state. Cell 141, 304-314. Métin, C., Vallee, R. B., Rakic, P. and Bhide, P. G. (2008). Modes and mishaps of neuronal migration in the mammalian brain. J. Neurosci. 28, 11746-11752. Millecamps, S. and Julien, J. P. (2013). Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 14, 161-176. Mitchell, D. J., Blasier, K. R., Jeffery, E. D., Ross, M. W., Pullikuth, A. K., Suo, D., Park, J., Smiley, W. R., Lo, K. W., Shabanowitz, J. et al. (2012). Trk activation of the ERK1/2 kinase pathway stimulates intermediate chain phosphorylation and recruits cytoplasmic dynein to signaling endosomes for retrograde axonal transport. J. Neurosci. 32, 15495-15510. Mokánszki, A., Körhegyi, I., Szabó, N., Bereg, E., Gergev, G., Balogh, E., Bessenyei, B., Sümegi, A., Morris-Rosendahl, D. J., Sztriha, L. et al. (2012). Lissencephaly and band heterotopia: LIS1, TUBA1A, and DCX mutations in Hungary. J. Child Neurol. 27, 1534-1540. Morfini, G., Pigino, G., Mizuno, N., Kikkawa, M. and Brady, S. T. (2007). Tau binding to microtubules does not directly affect microtubule-based vesicle motility. J. Neurosci. Res. 85, 2620-2630. Moughamian, A. J. and Holzbaur, E. L. (2012). Dynactin is required for transport initiation from the distal axon. Neuron 74, 331-343. Müller, M. J., Klumpp, S. and Lipowsky, R. (2008). Tug-of-war as a cooperative mechanism for bidirectional cargo transport by molecular motors. Proc. Natl. Acad. Sci. USA 105, 4609-4614. Münch, C., Sedlmeier, R., Meyer, T., Homberg, V., Sperfeld, A. D., Kurt, A., Prudlo, J., Peraus, G., Hanemann, C. O., Stumm, G. et al. (2004). Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 63, 724-726. Nakajima, K., Yin, X., Takei, Y., Seog, D. H., Homma, N. and Hirokawa, N. (2012). Molecular motor KIF5A is essential for GABA(A) receptor transport, and KIF5A deletion causes epilepsy. Neuron 76, 945-961. Nakata, T. and Hirokawa, N. (2003). Microtubules provide directional cues for polarized axonal transport through interaction with kinesin motor head. J. Cell Biol. 162, 1045-1055. Nakata, T., Niwa, S., Okada, Y., Perez, F. and Hirokawa, N. (2011). Preferential binding of a kinesin-1 motor to GTP-tubulin-rich microtubules underlies polarized vesicle transport. J. Cell Biol. 194, 245-255. Namba, T., Nakamuta, S., Funahashi, Y. and Kaibuchi, K. (2011). The role of selective transport in neuronal polarization. Dev. Neurobiol. 71, 445-457. Nanetti, L., Baratta, S., Panzeri, M., Tomasello, C., Lovati, C., Azzollini, J., Gellera, C., Di Bella, D., Taroni, F. and Mariotti, C. (2012). Novel and recurrent spastin mutations in a large series of SPG4 Italian families. Neurosci. Lett. 528, 42-45. Newsway, V., Fish, M., Rohrer, J. D., Majounie, E., Williams, N., Hack, M., Warren, J. D. and Morris, H. R. (2010). Perry syndrome due to the DCTN1 G71R mutation: a distinctive levodopa responsive disorder with behavioral syndrome, vertical gaze palsy, and respiratory failure. Mov. Disord. 25, 767-770. Niwa, S., Nakajima, K., Miki, H., Minato, Y., Wang, D. and Hirokawa, N. (2012). KIF19A is a microtubule-depolymerizing kinesin for ciliary length control. Dev. Cell 23, 1167-1175. Niwa, S., Takahashi, H. and Hirokawa, N. (2013). b-Tubulin mutations that cause severe neuropathies disrupt axonal transport. EMBO J. Ori-McKenney, K. M., Xu, J., Gross, S. P. and Vallee, R. B. (2010). A cytoplasmic dynein tail mutation impairs motor processivity. Nat. Cell Biol. 12, 1228-1234. Ou, C. Y., Poon, V. Y., Maeder, C. I., Watanabe, S., Lehrman, E. K., Fu, A. K., Park, M., Fu, W. Y., Jorgensen, E. M., Ip, N. Y. et al. (2010). Two cyclindependent kinase pathways are essential for polarized trafficking of presynaptic components. Cell 141, 846-858. Pandey, J. P. and Smith, D. S. (2011). A Cdk5-dependent switch regulates Lis1/Ndel1/ dynein-driven organelle transport in adult axons. J. Neurosci. 31, 17207-17219. Perlson, E., Maday, S., Fu, M. M., Moughamian, A. J. and Holzbaur, E. L. (2010). Retrograde axonal transport: pathways to cell death? Trends Neurosci. 33, 335-344. Pfister, K. K., Shah, P. R., Hummerich, H., Russ, A., Cotton, J., Annuar, A. A., King, S. M. and Fisher, E. M. (2006). Genetic analysis of the cytoplasmic dynein subunit families. PLoS Genet. 2, e1. Pilling, A. D., Horiuchi, D., Lively, C. M. and Saxton, W. M. (2006). Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol. Biol. Cell 17, 2057-2068. Poirier, K., Saillour, Y., Bahi-Buisson, N., Jaglin, X. H., Fallet-Bianco, C., Nabbout, R., Castelnau-Ptakhine, L., Roubertie, A., Attie-Bitach, T., Desguerre, I. et al. (2010). Mutations in the neuronal b-tubulin subunit TUBB3 result in malformation of Journal of Cell Science Transport and disease cortical development and neuronal migration defects. Hum. Mol. Genet. 19, 44624473. Puls, I., Jonnakuty, C., LaMonte, B. H., Holzbaur, E. L., Tokito, M., Mann, E., Floeter, M. K., Bidus, K., Drayna, D., Oh, S. J. et al. (2003). Mutant dynactin in motor neuron disease. Nat. Genet. 33, 455-456. Puls, I., Oh, S. J., Sumner, C. J., Wallace, K. E., Floeter, M. K., Mann, E. A., Kennedy, W. R., Wendelschafer-Crabb, G., Vortmeyer, A., Powers, R. et al. (2005). Distal spinal and bulbar muscular atrophy caused by dynactin mutation. Ann. Neurol. 57, 687-694. Qiu, W., Derr, N. D., Goodman, B. S., Villa, E., Wu, D., Shih, W. and ReckPeterson, S. L. (2012). Dynein achieves processive motion using both stochastic and coordinated stepping. Nat. Struct. Mol. Biol. 19, 193-200. Reck-Peterson, S. L., Yildiz, A., Carter, A. P., Gennerich, A., Zhang, N. and Vale, R. D. (2006). Single-molecule analysis of dynein processivity and stepping behavior. Cell 126, 335-348. Reid, E., Kloos, M., Ashley-Koch, A., Hughes, L., Bevan, S., Svenson, I. K., Graham, F. L., Gaskell, P. C., Dearlove, A., Pericak-Vance, M. A. et al. (2002). A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10). Am. J. Hum. Genet. 71, 1189-1194. Rolls, M. M. (2011). Neuronal polarity in Drosophila: sorting out axons and dendrites. Dev. Neurobiol. 71, 419-429. Romaniello, R., Tonelli, A., Arrigoni, F., Baschirotto, C., Triulzi, F., Bresolin, N., Bassi, M. T. and Borgatti, R. (2012). A novel mutation in the b-tubulin gene TUBB2B associated with complex malformation of cortical development and deficits in axonal guidance. Dev. Med. Child Neurol. 54, 765-769. Sabo, S. L. and McAllister, A. K. (2003). Mobility and cycling of synaptic proteincontaining vesicles in axonal growth cone filopodia. Nat. Neurosci. 6, 1264-1269. Salinas, S., Bilsland, L. G. and Schiavo, G. (2008). Molecular landmarks along the axonal route: axonal transport in health and disease. Curr. Opin. Cell Biol. 20, 445453. Saxena, S. and Caroni, P. (2007). Mechanisms of axon degeneration: from development to disease. Prog. Neurobiol. 83, 174-191. Saxton, W. M. and Hollenbeck, P. J. (2012). The axonal transport of mitochondria. J. Cell Sci. 125, 2095-2104. Schaefer, M. K., Schmalbruch, H., Buhler, E., Lopez, C., Martin, N., Guénet, J. L. and Haase, G. (2007). Progressive motor neuronopathy: a critical role of the tubulin chaperone TBCE in axonal tubulin routing from the Golgi apparatus. J. Neurosci. 27, 8779-8789. Schlager, M. A. and Hoogenraad, C. C. (2009). Basic mechanisms for recognition and transport of synaptic cargos. Mol. Brain 2, 25. Schliwa, M. and Woehlke, G. (2003). Molecular motors. Nature 422, 759-765. Shemesh, O. A., Erez, H., Ginzburg, I. and Spira, M. E. (2008). Tau-induced traffic jams reflect organelles accumulation at points of microtubule polar mismatching. Traffic 9, 458-471. Shubeita, G. T., Tran, S. L., Xu, J., Vershinin, M., Cermelli, S., Cotton, S. L., Welte, M. A. and Gross, S. P. (2008). Consequences of motor copy number on the intracellular transport of kinesin-1-driven lipid droplets. Cell 135, 1098-1107. Siththanandan, V. B. and Sellers, J. R. (2011). Regulation of myosin 5a and myosin 7a. Biochem. Soc. Trans. 39, 1136-1141. Song, A. H., Wang, D., Chen, G., Li, Y., Luo, J., Duan, S. and Poo, M. M. (2009). A selective filter for cytoplasmic transport at the axon initial segment. Cell 136, 11481160. Soppina, V., Rai, A. K., Ramaiya, A. J., Barak, P. and Mallik, R. (2009). Tug-of-war between dissimilar teams of microtubule motors regulates transport and fission of endosomes. Proc. Natl. Acad. Sci. USA 106, 19381-19386. Stiess, M. and Bradke, F. (2011). Neuronal polarization: the cytoskeleton leads the way. Dev. Neurobiol. 71, 430-444. Stockmann, M., Meyer-Ohlendorf, M., Achberger, K., Putz, S., Demestre, M., Yin, H., Hendrich, C., Linta, L., Heinrich, J., Brunner, C. et al. (2012). The dynactin p150 subunit: cell biology studies of sequence changes found in ALS/MND and Parkinsonian Syndromes. J. Neural Transm. 120, 785-798. Tanaka, T., Serneo, F. F., Higgins, C., Gambello, M. J., Wynshaw-Boris, A. and Gleeson, J. G. (2004). Lis1 and doublecortin function with dynein to mediate coupling of the nucleus to the centrosome in neuronal migration. J. Cell Biol. 165, 709-721. Teuling, E., van Dis, V., Wulf, P. S., Haasdijk, E. D., Akhmanova, A., Hoogenraad, C. C. and Jaarsma, D. (2008). A novel mouse model with impaired dynein/dynactin function develops amyotrophic lateral sclerosis (ALS)-like features in motor neurons and improves lifespan in SOD1-ALS mice. Hum. Mol. Genet. 17, 2849-2862. Tischfield, M. A., Baris, H. N., Wu, C., Rudolph, G., Van Maldergem, L., He, W., Chan, W. M., Andrews, C., Demer, J. L., Robertson, R. L. et al. (2010). Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell 140, 74-87. 2329 Tischfield, M. A., Cederquist, G. Y., Gupta, M. L., Jr and Engle, E. C. (2011). Phenotypic spectrum of the tubulin-related disorders and functional implications of disease-causing mutations. Curr. Opin. Genet. Dev. 21, 286-294. Toba, S., Watanabe, T. M., Yamaguchi-Okimoto, L., Toyoshima, Y. Y. and Higuchi, H. (2006). Overlapping hand-over-hand mechanism of single molecular motility of cytoplasmic dynein. Proc. Natl. Acad. Sci. USA 103, 5741-5745. Tsai, J. W., Bremner, K. H. and Vallee, R. B. (2007). Dual subcellular roles for LIS1 and dynein in radial neuronal migration in live brain tissue. Nat. Neurosci. 10, 970979. Uribe, V. (2010). DCTN1 mutations are implicated in multiple neurodegenerative disorders. Clin. Genet. 77, 32-34. Vale, R. D. (2003). The molecular motor toolbox for intracellular transport. Cell 112, 467-480. Vale, R. D., Schnapp, B. J., Mitchison, T., Steuer, E., Reese, T. S. and Sheetz, M. P. (1985). Different axoplasmic proteins generate movement in opposite directions along microtubules in vitro. Cell 43, 623-632. Vale, R. D., Funatsu, T., Pierce, D. W., Romberg, L., Harada, Y. and Yanagida, T. (1996). Direct observation of single kinesin molecules moving along microtubules. Nature 380, 451-453. Vallee, R. B., Seale, G. E. and Tsai, J. W. (2009). Emerging roles for myosin II and cytoplasmic dynein in migrating neurons and growth cones. Trends Cell Biol. 19, 347355. van Spronsen, M., Mikhaylova, M., Lipka, J., Schlager, M. A., van den Heuvel, D. J., Kuijpers, M., Wulf, P. S., Keijzer, N., Demmers, J., Kapitein, L. C. et al. (2013). TRAK/Milton motor-adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron 77, 485-502. Verhey, K. J. and Hammond, J. W. (2009). Traffic control: regulation of kinesin motors. Nat. Rev. Mol. Cell Biol. 10, 765-777. Vershinin, M., Carter, B. C., Razafsky, D. S., King, S. J. and Gross, S. P. (2007). Multiple-motor based transport and its regulation by Tau. Proc. Natl. Acad. Sci. USA 104, 87-92. Watanabe, K., Al-Bassam, S., Miyazaki, Y., Wandless, T. J., Webster, P. and Arnold, D. B. (2012). Networks of polarized actin filaments in the axon initial segment provide a mechanism for sorting axonal and dendritic proteins. Cell Rep. 2, 1546-1553. Weedon, M. N., Hastings, R., Caswell, R., Xie, W., Paszkiewicz, K., Antoniadi, T., Williams, M., King, C., Greenhalgh, L., Newbury-Ecob, R. et al. (2011). Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease. Am. J. Hum. Genet. 89, 308-312. Welte, M. A. (2004). Bidirectional transport along microtubules. Curr. Biol. 14, R525R537. Willemsen, M. H., Vissers, L. E., Willemsen, M. A., van Bon, B. W., Kroes, T., de Ligt, J., de Vries, B. B., Schoots, J., Lugtenberg, D., Hamel, B. C. et al. (2012). Mutations in DYNC1H1 cause severe intellectual disability with neuronal migration defects. J. Med. Genet. 49, 179-183. Witte, H. and Bradke, F. (2008). The role of the cytoskeleton during neuronal polarization. Curr. Opin. Neurobiol. 18, 479-487. Wong, M. Y., Zhou, C., Shakiryanova, D., Lloyd, T. E., Deitcher, D. L. and Levitan, E. S. (2012). Neuropeptide delivery to synapses by long-range vesicle circulation and sporadic capture. Cell 148, 1029-1038. Wynshaw-Boris, A., Pramparo, T., Youn, Y. H. and Hirotsune, S. (2010). Lissencephaly: mechanistic insights from animal models and potential therapeutic strategies. Semin. Cell Dev. Biol. 21, 823-830. Yamada, K., Andrews, C., Chan, W. M., McKeown, C. A., Magli, A., de Berardinis, T., Loewenstein, A., Lazar, M., O’Keefe, M., Letson, R. et al. (2003). Heterozygous mutations of the kinesin KIF21A in congenital fibrosis of the extraocular muscles type 1 (CFEOM1). Nat. Genet. 35, 318-321. Yi, J. Y., Ori-McKenney, K. M., McKenney, R. J., Vershinin, M., Gross, S. P. and Vallee, R. B. (2011). High-resolution imaging reveals indirect coordination of opposite motors and a role for LIS1 in high-load axonal transport. J. Cell Biol. 195, 193-201. Yildiz, A., Tomishige, M., Vale, R. D. and Selvin, P. R. (2004). Kinesin walks handover-hand. Science 303, 676-678. Yildiz, A., Tomishige, M., Gennerich, A. and Vale, R. D. (2008). Intramolecular strain coordinates kinesin stepping behavior along microtubules. Cell 134, 1030-1041. Yuan, A., Kumar, A., Peterhoff, C., Duff, K. and Nixon, R. A. (2008). Axonal transport rates in vivo are unaffected by tau deletion or overexpression in mice. J. Neurosci. 28, 1682-1687. Zhao, C., Takita, J., Tanaka, Y., Setou, M., Nakagawa, T., Takeda, S., Yang, H. W., Terada, S., Nakata, T., Takei, Y. et al. (2001). Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell 105, 587-597. Zyłkiewicz, E., Kijańska, M., Choi, W. C., Derewenda, U., Derewenda, Z. S. and Stukenberg, P. T. (2011). The N-terminal coiled-coil of Ndel1 is a regulated scaffold that recruits LIS1 to dynein. J. Cell Biol. 192, 433-445.