Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
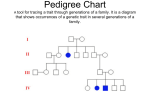
Indian J. Pharm. Biol. Res. 2014; 2(4):68-76 CODEN (USA): IJPB07 ISSN: 2320-9267 Indian Journal of Pharmaceutical and Biological Research (IJPBR) Journal homepage: www.ijpbr.in Review Article Recent trends and advances in hemophilia – its management and new therapeutic outcomes Jiguru Prasant* Flat no: 7, 4th Floor, Sonaappartments, 50 sebastain road, Rezimental Bazar, Secunderabad, India ARTICLE INFO: Article history: Received: 23 October 2014 Received in revised form: 5 November 2014 Accepted: 15 November 2014 Available online: 31 December 2014 Keywords: Hemophilia, Factor VIII, factor IX, Hemostasis, Thrombin. ABSTRACT Hemophilia unfortunately a less attracted disease for researchers compared to other life threatening diseases. The prevalence of hemophilia is estimated to be about 1:10,000 birth and that of the severe form of the disease to be about 6% per 1,00,000 population. The most pathetic part of this disease is that even medical personnel are sometimes not familiar with its diagnosis and management. There is obviously a need to establish facilities and treatment options that will help the patient with hemophilia to manage their life with ease. As this is a genetic disorder no complete cure is possible as of now. The only available treatment option is the infusion of factors and some adjuvant therapies depending upon the bleeding conditions .The initiative for the development of new dosage forms, new delivery systems of the existing therapies or new treatment options has to be driven by pharmacy professionals. This article present an overview of hemophilia, in order to drag the attention of medical as well as pharmacy professionals for the benefit of millions of hemophilic patients . Introduction Hemophilia is a group of inherited blood disorders in which the blood does not clot properly. Hemophilia is the standard international spelling, also known as haemophilia in the UK, other translations include: hémophilie, hemofilie, hemofili, hemofilia, hämophilie, emofilia. We will use the standard international spelling for the purpose of this section[1]. Bleeding disorders are due to defects in the blood vessels, the coagulation mechanism, or the blood platelets[2]. An affected individual may bleed spontaneously or for longer than a healthy person after injury or surgery. The blood coagulation mechanism is a process which transforms the blood from a liquid into a solid, and involves several different clotting factors. The mechanism generates fibrin when it is activated, which together with the platelet plug, stops the bleeding. When coagulation factors are missing or deficient the blood does not clot properly and bleeding continues. Patients with Hemophilia A or B have a genetic defect which results in a deficiency in one of the blood clotting factors. * Types of Hemophilia / Haemophilia Hemophilia A and Hemophilia B[3] There are two main types of hemophilia - Hemophilia A (due to factor VIII deficiency) and Hemophilia B (due to factor IX deficiency). They are clinically almost identical and are associated with spontaneous bleeding into joints and muscles and internal or external bleeding after injury or surgery. After repeated bleeding episodes permanent damage may be caused to the joints and muscles that have been affected, particularly the ankles, knees and elbows[4]. Approximately 1 in 5,000 males is born with Hemophilia A, and 1 in 30,000 males is born with Hemophilia B. Hemophilia affects people of all races and ethnic origins globally. The conditions are both X-linked and virtually all sufferers of hemophilia are males. Female carriers may also bleed abnormally, because some have low levels of the relevant clotting factor. People with hemophilia have a genetic mutation in the affected gene on the X chromosome, which results in reduced production of Factor VIII or IX and creates a bleeding Corresponding Author: Jiguru Prasant, Flat no: 7, 4th Floor, Sonaappartments, 50 sebastain road, Rezimental Bazar, Secunderabad, India. E-Mail: [email protected] 68 Prasant et al. / Indian J. Pharm. Biol. Res., 2014; 2(4):68-76 tendency, because coagulation takes much longer than normal, thus making the clot weak and unstable[5]. Approximately one third of patients with hemophilia have no family history of the disease, either because of new genetic mutations, or because previous affected generations either had daughters (who were carriers) or sons who died in early childhood from hemophilia or any other cause or who were not affected[6,7] [6,7]. Acquired hemophilia This is very rare. The patient develops the condition during his/her lifetime and it does not have a genetic or heritable cause. It occurs when the bodyy forms antibodies that attack one or more blood clotting factors, (usually factor VIII), thus preventing the blood clotting mechanism from working properly. Patients may be male or female and the pattern of bleeding is rather different from that of classical classi hemophilia, the joints being rarely affected. The disorder is particularly associated with old age and occasionally complicates pregnancy[8]. Causes Hemophilia[9] People with hemophilia are born with it. It is caused by a fault in one of the genes thatt determine how the body makes blood clotting factor VIII or IX. These genes are located on the X chromosome. To understand how hemophilia is inherited, it is important to learn about chromosomes. Female (X + Xfaulty) is a carrier, but does not have hemophilia. The “good” X chromosome allows the production of enough clotting factor to prevent serious bleeding problems. Male (Y + Xfaulty) will develop hemophilia and can pass it on. If the father has hemophilia and the mother has no faulty gene (is not a carrier): Father (Y + Xfaulty). Mother (X + X). Review Article What are chromosomes? Chromosomes are blocks of DNA (deoxyribonucleic ibonucleic acid). They contain very detailed and specific instructions that determine: How the cells in a baby's body develop. What features the baby will have, including, for example, hair and eye color. Whether the baby is male or female. In humans there are 23 pairs of chromosomes, including the sex chromosome pair. There are two types of sex chromosome: The X chromosome The Y chromosome All humans have a pair of sex chromosomes: [10] Males have an X + Y pair Females have an X + X pair NB Females do not have any Y chromosomes. What chromosomes do we inherit from our parents? A Male inherits his X chromosome from his mother Y chromosome from his father A Female inherits One X chromosome from her mother One X chromosome from her father She does not inherit erit both X chromosomes from her mother. She has no Y chromosomes. [11,12]How can we calculate the risk of hemophilia in offspring? x-linked linked recessive, carrier mother (Before reading on, remember that the faulty gene is never on the Y chromosome. If it is present, it will be on the X chromosome.) There is no risk of inherited hemophilia in their sons because becaus boys will inherit their X chromosome from the mother, not the father (they inherit the father's Y chromosome only, which does not have the faulty gene). All the daughters will be carriers but will not develop hemophilia although they will inherit the father's fat X chromosome, which has the faulty gene. However, their maternal X chromosome, which does not have the faulty gene, 69 Prasant et al. / Indian J. Pharm. Biol. Res., 2014; 2(4):68-76 usually allows the production of enough clotting factor to prevent serious bleeding problems. If the father does not have hemophilia and the mother has a faulty gene: Father (Y + X). Mother (X + Xfaulty). There is a 50% chance that sons will develop hemophilia because: There is a 50% risk that a son will inherit his mother's Xfaulty chromosome, plus his father's Y chromosome - he will have hemophilia. There is a 50% chance he will inherit his mother's "good" X chromosome, plus his father's Y chromosome - he will not have hemophilia. There is a 50% chance that daughters will be carriers, (but no chance of developing hemophilia), because: There is a 50% chance she will inherit her mother's Xfaulty chromosome, making her a carrier. There is a 50% chance she will inherit her mother's "good" X chromosome, which would mean she would not be a carrier. Approximately one third of patients with hemophilia have no family history of the disease, either because of new genetic mutations, or because previous affected generations either had daughters (who were carriers) or sons who died in early childhood from hemophilia or any other cause or who were not affected. Pathophysiology[13-15] Factor VIII production, processing, and structure Primary sites of factor VIII (FVIII) production are thought to be the liver and the reticuloendothelial system. Liver transplantation corrects FVIII deficiency in persons with hemophilia, and persons with mild hemophilia with progressive liver disease have a rise in FVIII levels, thus establishing the liver as the major site of FVIII synthesis. FVIII messenger RNA has been detected in the liver, spleen, and other tissues.[3] Studies of FVIII production in transfected cell lines have shown that following synthesis, FVIII moves to the lumen of the endoplasmic reticulum, where it is bound to several proteins that regulate secretion, particularly immunoglobulin binding protein, from which it has to dissociate in an energy-dependent process. Cleavage of FVIII's signal peptide and the addition of oligosaccharides also occur in the endoplasmic reticulum. The chaperone proteins, calnexin and calreticulin, enhance both FVIII secretion and degradation. A part of the factor FVIII protein in the endoplasmic reticulum is degraded within the cell. The other part enters the Golgi apparatus, where several changes occur to produce the heavy and light chains and to modify the carbohydrates. The addition of sulfates to tyrosine residues of the heavy and light chains is necessary for full procoagulant activity, with the sulfated region playing a role in thrombin interaction. This posttranslational sulfation of tyrosine residues impacts the procoagulant activity of factor VIII and its interaction with von Willebrand factor (vWF). vonWillebrand factor[16] FVIII circulates in plasma in a noncovalently bound complex with vWF, which plays significant roles in the function, production, stabilization, conformation, and immunogenicity of FVIII.[4] VWF has been termed FVIII-related antigen (FVIII-R); related terminology for FVIII is FVIII-coagulant (FVIII-C). VWF appears to promote assembly of the heavy and light chains of FVIII and more efficient secretion of FVIII from the endoplasmic reticulum. It also directs FVIII into the WeibelPalade bodies, which are the intracellular storage sites for vWF. In plasma, vWF stabilizes FVIII and protects it from degradation. In the presence of normal vWF protein, the halflife of FVIII is approximately 12 hours, whereas in the absence of vWF, the half-life of FVIII-C is reduced to 2 hours.[5, 6, 7] The clotting cascade [17]The role of the coagulation system is to produce a stable fibrin clot at sites of injury. The clotting mechanism has 2 pathways: intrinsic and extrinsic. See the image below. Coagulation pathway The intrinsic system is initiated when factor XII is activated by contact with damaged endothelium.[18] The activation of factor XII can also initiate the extrinsic pathway, fibrinolysis, kinin generation, and complement activation. Review Article In conjunction with high-molecular-weight kininogen (HMWK), factor XIIa converts prekallikrein (PK) to kallikrein and activates factor XI. Activated factor XI, in turn, activates factor IX in a calcium-dependent reaction. Factor IXa can bind phospholipids. Then, factor X is activated on the cell surface; 70 Prasant et al. / Indian J. Pharm. Biol. Res., 2014; 2(4):68-76 activation of factor X involves a complex (tenase complex) of factor IXa, thrombin-activated FVIII, calcium ions, and phospholipid. In the extrinsic system, [19]. The conversion of factor X to factor Xa involves tissue factor (TF), or thromboplastin; factor VII; and calcium ions. TF is released from the damaged cells and is thought to be a lipoprotein complex that acts as a cell surface receptor for factor VII, with its resultant activation. TF also adsorbs factor X to enhance the reaction between factor VIIa, factor X, and calcium ions. Factor IXa and factor XII fragments can also activate factor VII. In the common pathway, factor Xa (generated through the intrinsic or extrinsic pathways) forms a prothrombinase complex with phospholipids, calcium ions, and thrombin- activated factor Va. The complex cleaves prothrombin into thrombin andprothrombin fragments 1 and 2. Thrombin converts fibrinogen into fibrin and activates FVIII, factor V, and factor XIII. Fibrinopeptides A and B, the results of the cleavage of peptides A and B by thrombin, cause fibrin monomers to form and then polymerize into a meshwork of fibrin; the resultant clot is stabilized by factor XIIIa and the cross-linking of adjacent fibrin strands. Because of the complex interactions of the intrinsic and extrinsic pathways (factor IXa activates factor VII), the existence of only one in vivo pathway with different mechanisms of activation has been suggetdSee the image below. The hemostatic pathway. APC = activated protein C The hemostatic pathway. APC = activated protein C (APC); AT-III = antithrombin III; FDP = fibrin degradation products; HC-II = heparin cofactor II; HMWK = high-molecular-weight kininogen; PAI = plasminogen activator inhibitor; sc-uPA = single-chain urokinase plasminogen activator; tc-uPA = twochain urokinase plasminogen activator; TFPI = tissue factor pathway inhibitor; tPA = tissue plasminogen activator [20]FVIII and factor IX circulate in an inactive form. When activated, these 2 factors cooperate to cleave and activate factor X, a key enzyme that controls the conversion of fibrinogen to fibrin. Therefore, the lack of FVIII may significantly alter clot formation and, as a consequence, result in clinical bleeding. Three levels of hemophilia are recognized, according to the level of clotting factor amounts in the blood. These are often expressed as percentages of normal: Above 5% - mild hemophilia 1% to 5% - moderate hemophilia Less than 1% - severe hemophilia Hemophilia Symptoms and Diagnosis[21,22] Hemophilia symptoms vary, depending on the degree of blood clotting factor (coagulation factor) deficiency and they also depend on the nature of any injury. Moderate hemophilia Those with inherited moderate hemophilia will be noticeable early on. The child will bruise easily and may also experience internal bleeding symptoms, especially around the joints, and after a blow or a fall. Bleeding that occurs inside a joint is usually referred to as a joint bleed. Review Article Mild hemophilia People with inherited mild hemophilia may not have any symptoms until an event occurs which wounds the skin or tissue, such as a dental procedure or surgery, and results in prolonged bleeding. In societies where male circumcision is carried out soon after birth, mild hemophilia will be detected earlier. Joint bleeding is uncommon. 71 Prasant et al. / Indian J. Pharm. Biol. Res., 2014; 2(4):68-76 Symptoms of a joint bleed Tingling sensation in the joint, Pain in the joint, Irritation in the joint, If left untreated, the patient may eventually experience: More severe pain in the joint, Joint stiffness, The affected area becomes swollen, tender and hot Joint bleeds most commonly affect the: Ankles, Knees, Elbows and may less commonly affect the shoulders, hips or other joints. Any surgical intervention, circumcision, dental procedure or injury will result in prolonged bleeding in a person with hemophilia. Symptoms are similar to those found in moderate hemophilia, but occur more frequently and are usually more severe. A child with severe hemophilia will often bleed for no apparent reason, often referred to as spontaneous bleeding. Most commonly, in early childhood from about 18 months of age, the nose or mouth start to bleed or apparently spontaneous bruises appear, particularly on the legs. Parents are sometimes suspected of causing non-accidental injury (deliberate harm) to their children. Symptoms of hemophilia type bleeding may include:[23,34,25] Several large or deep bruises, Joint pain or swelling, Unexplained bleeding or bruising, Blood in feces (stools), Blood in urine, Unexplained nosebleeds, Unexplained gum bleeding, Tightness in the joints Intracranial hemorrhage (bleeding inside the skull) About 1 in every 30 patients with hemophilia will have intracranial hemorrhage at least once during their lives. This should be treated as a medical emergency. Spontaneous intracranial hemorrhage is rare and in many cases bleeding inside the skull will be the result of a blow to the head. Symptoms of intracranial hemorrhage include: A bad headache, Vomiting, Confusion, Fitting (Convulsion), Loss of balance, Slurred speech, or other speaking difficulties, Stiff neck, Vision problems, Loss of coordination, Some of the facial muscles do not work (sometimes all of them) How is hemophilia diagnosed? Prenatal testing - if a pregnant woman has a history of hemophilia, a hemophilia gene test can be done during pregnancy. A sample of placenta is removed from the uterus and tested. This test is known as a CVS (chorionic villus sampling) test. Blood test - if a doctor suspects a child may have hemophilia a blood test can determine whether the patient has hemophilia A or B, and how severe it is. Blood tests can be performed from the time of birth onwards. Treatment for Hemophilia / Haemophilia[26,27] Up to a few decades ago a considerable proportion of patients with hemophilia died prematurely because of hemophilia. Tragically, many deaths were the result of childhood injury or Review Article surgery. Over the last forty years treatment has advanced so much that the vast majority of patients today are expected to live long and active lives. The main breakthrough in treatment occurred when coagulation factor deficiencies linked to hemophilia could be identified and then replaced, using products derived from human blood. In the past patients used to receive whole blood or plasma infusions to control episodes of bleeding. Even though this helped, levels of clotting factors, especially factors VIII and IX, never reached the levels required for really effective blood coagulation, nor could these levels be sustained - in other words, serious bleeding was only partly treated. Cryoprecipitate, made through the cold precipitation of frozen plasma from1965 onwards, was the first really effective treatment for hemophilia A. Freeze-dried concentrates made from human plasma containing the right levels of Factors VIII and IX became available in the late 1960s and early 1970s. Being able to keep the treatment at home and use it as required meant that patients could travel, leave the home, go to work, and enjoy a level of independence. However, a large number of patients subsequently became infected with blood-borne pathogens, such as hepatitis B, hepatitis C and HIV. From the mid 1980s rigorous donor selection and viral inactivation procedures reduced the risk of blood-borne viral transmission to nearly zero. During the 1990s it became possible to prepare synthetic (recombinant) factors, using specially prepared mammalian cells and these recombinant concentrates are now widely used. Hemophilia treatment will mainly depend on its severity and for patients with Hemophilia A or B involves clotting factor replacement therapy. There are two approaches: • On demand - giving treatment to stop prolonged bleeding when it occurs. This is more common in the management of patients with mild hemophilia. • Preventative treatment (prophylaxis) - medication to prevent bleeding episodes, and subsequent complications, such as joint and/or muscle damage. More commonly used for patients with moderate or severe hemophilia. Clotting factor concentrates [28] Clotting factor concentrates can be made in two different ways: Plasma-derived clotting factors - prepared from the plasma of donated human blood. Recombinant clotting factors - the first generation of recombinant products use animal products in the culture medium and had human albumin (a human blood product) added as a stabiliser. Second generation products use animalderived materials in the culture medium but do not have added albumin and instead use sucrose or other non-human derived material as a stabiliser. Third generation clotting factors have no albumin present at any stage of their preparation. Mouse monoclonal antibodies have been routinely used in the purification of coagulation factors for many years but a recently licensed recombinant factor VIII employs a synthetic 72 Prasant et al. / Indian J. Pharm. Biol. Res., 2014; 2(4):68-76 ligand for this step. [29]This has resulted in the production of the first factor VIII concentrate to be free of all exogenous human and animal protein, a goal which was reached for hemophilia B when the first recombinant factor IX was licensed in 1997. Desmopressin (DDAVP)(for mild hemophilia A)[30] This medication is a synthetic hormone which encourages the body to produce more of its own Factor VIII. It is unsuitable for patients with hemophilia B and those with severe hemophilia A. In patients with milder forms of hemophilia A, factor VIII replacement therapy may be necessary, especially for severe bleeds, or after serious injury or major surgery. RICE (Rest, Ice, Compression, Elevation)[31,32,33] RICE is a treatment many health care professionals recommend for joint bleeds. It also reduces swelling and tissue damage when used together with clotting factor concentrates. Administering clotting factor concentrates The medication is injected into a vein - generally in the back of the hand or at the crook of the elbow. Initial treatments are usually administered by a doctor or nurse at a hospital or clinic. Most adults can learn how to do this themselves, which means they can stop bleeding rapidly and effectively wherever they are. If the patient is a child the parents or caregivers (UK/Ireland/Australia: carers) can learn how to administer treatment. The majority of very young patients can receive most of their treatment at home. If a patient is finding it hard to access a suitable vein, or if intensive treatment is required, a port-a-cath, or an external catheter called a Broviac or Hickman line can be placed surgically into a vein, allowing factor replacement therapies to be given, and blood to be drawn easily for routine emergency tests. The use of such catheters can be complicated by infection and blockage and they have to be used with great care. Treating bleeds[34] Bleeding episodes (bleeds) are an inevitable complication for patients with hemophilia A and B, even for patients with mild forms. As the underlying problem is one of prolonged bleeding, rather than rapid bleeding, they often appear not to be medical emergencies. If a person with hemophilia experiences any of the following he should seek immediate skilled medical help:[35-37] There is an injury to the neck, mouth, tongue, face or eye. There is a severe blow to the head. Bleeding is heavy or persistent. There is severe pain or swelling in any part of the body. An open wound requires stitching. Most other bleeds, such as joint/muscle bleeds, small injuries and cuts that do not require stitches, and nosebleeds are generally treated at home, but patients should always seek the advice of a healthcare professional when in doubt. Any treatment will be more effective if it is started early. Review Article Storing treatment[38,39] Factor concentrates should usually be stored in a refrigerator but are stable at room temperature for quite long periods. They should not be frozen as this may damage the vials or syringes. Some may be taken out for travel but should ideally be kept in a cool bag. Read instructions on product storage. If you are unsure, check with a health care professional or qualified pharmacist. Inhibitors[40-44] Approximately 30% of people with severe hemophilia A develop antibodies to transfused factor VIII, usually shortly after their first few treatments. These antibodies (also called inhibitors) prevent the factor VIIII treatment working properly. It is often the case that, after a while, the inhibitors disappear and only about 10% or less of people with severe hemophilia A will suffer from long term inhibitors. In recent years it has become possible to prevent inhibitors becoming persistent through immune tolerance induction therapy. Where inhibitors do not respond to this approach alternative treatments are available. Inhibitors rarely develop in mild hemophilia A or in hemophilia B of any severity. Prevention[45-50] The transmittance of the hemophilia to the next generation can be prevented by the following methods. 1)Prenatal intrauterine diagnosis with termination of pregnancy as an option 2)Pre implantation genetic diagnostic testing (PGD) 3)IVF with egg/sperm donation therapies under investigation Longer acting factor concentrates[51,52,53] A longer-acting factor VIII concentrate has recently been approved by the FDA for clinical trials. The hope is that one infusion a week rather than three to four infusions a week will provide prophylaxis against spontaneous bleeding. A preparation in which recombinant factor IX is fused to a portion of the immunoglobulin Fc protein shows prolonged survival and efficacy in animal models.29 Clinical trials are planned but have not yet been initiated. Gene therapy Scientists at The Children's Hospital of Philadelphia conducted an experimental protocol involving dogs with a genetic defect predisposing them to develop hemophilia B. Functional genes capable of producing the clotting Factor IX were attached to an adeno-associated virus vector and injected into the leg muscles of the animals. The theory was that the genetically modified virus would carry the missing gene to muscle cells, which in turn would take up the gene and begin producing the clotting factor and releasing it into the blood stream. Sustained therapeutic expression of factors VIII and IX has been achieved in preclinical studies using a wide range of 73 Prasant et al. / Indian J. Pharm. Biol. Res., 2014; 2(4):68-76 gene transfer technologies targeted at different tissues. This achievement has led to six different phase I/II clinical trials that resulted in limited efficacy but minimal toxicity. Recombinant adeno-associated viral vectors appear most promising for hemophilia gene therapy. Attempts are being made to learn more about the immunology of inhibitors and ways to prevent them or improve the success rate of immune tolerance. Future[54,55] Gene therapy, oral delivery of factor concentrates, long acting infusions, controlled release implants, prolonged release of other adjuvant therapy drugs are the areas to be concentrated for making the life of hemophilic patients easier. A schematic representation is given in the Figure 7. Conclusion Hemophilia is a bleeding disorder, very hard to live with. As the number of patients reported with hemophilia is comparatively less than other major diseases like cancer, cardiac diseases, and diabetics it seems to be less concentrated area by researchers. As technologies develop, the days are not far away when hemophilia is treated with ease and even complete cure possible. Conflict of interest statement We declare that we have no conflict of interest. Acknowledgement I would like to acknowledge the people who mean world to me, my parents, & my sister. I extend my respect to my parents and all elders to me in the family. I don’t imagine a life without their love and blessings. Thank you mom, dad, for showing faith in me and giving me liberty to choose what I desired. I consider myself the luckiest in the world to have such a supportive family, standing behind me with their love and support. References 12. 1. Peyvandi F, Jayandharan G, Chandy M, Srivastava A, Nakaya SM, et al. Genetic diagnosis of haemophilia and other inherited bleeding disorders. Haemophilia 12 Suppl 3: 82-89. 2. Mannucci PM,; Duga S, Peyvandi F, Recessively inherited coagulation disorders. Blood 2004;104:12431252. 3. Gitschier J, Wood WI, Goralka TM, Wion KL, Chen EY, et al. Characterization of the human factor VIII gene. Nature 1984;312: 326-330. 4. Yoshitake S, Schach BG, Foster DC, Davie EW, Kurachi K. Nucleotide sequence of the gene for human factor IX (antihemophilic factor B) Biochemistry 1985;24: 3736-3750. 5. Drayna D, White R (1985) The genetic linkage map of the human X chromosome. Science 230: 753-758. 6. Hoffman M . Blood Rev 2003;17 Suppl 1: S1-S5. 7. Ahmad SS, London FS, Walsh PN. The assembly of the factor X-activating complex on activated human platelets. J ThrombHaemost 2003;1: 48 8. O'Mahoney B (2002) Global haemophilia care challenge and opportunities: World Federation of Hemophilia 9. Peake IR, Lillicrap DP, Boulyjenkov V, Briet E, Chan V, et al. Haemophilia: strategies for carrier detection and prenatal diagnosis. Bull World Health Organ 1993;71: 429-458. 10. Leuer M, Oldenburg J, Lavergne JM, Ludwig M, Fregin A, et al. Somatic mosaicism in hemophilia A: a fairly common event. Am J Hum Genet 2001;69: 75-87. 11. Jayandharan G, Shaji RV, Baidya S, Nair SC, Chandy M, et al. Identification of factor VIII gene mutations in 101 patients with haemophilia A: mutation analysis by inversion screening and multiplex PCR and CSGE and Review Article 13. 14. 15. 16. 17. 18. 19. 20. molecular modelling of 10 novel missense substitutions. Haemophilia 2005;11: 481- 491. Pavlova A, Brondke H, Musebeck J, Pollmann H, Srivastava A, et al. Molecular mechanisms underlying hemophilia A phenotype in seven females. J ThrombHaemost 2009;7: 976-982. White GC II, Rosendaal F, Aledort LM, Lusher JM, Rothschild C, Ingerslev J. Definitions in hemophilia: recommendation of the scientific subcommittee on factor VIII and factor IX of the scientific and standardization committee of the International Society on Thrombosis and Haemostasis. ThrombHaemost 2001;85: 560. Bolton-Maggs PH, Pasi KJ. Haemophilias A and B. Lancet 2003;361: 1801- 1809. Street AM, Ljung R, Lavery SA. Management of carriers and babies with haemophilia. Haemophilia 2008;3: 181-187. Schramm W, Royal S, Kroner B, Berntorp E, Giangrande P, et al. Clinical outcomes and resource utilization associated with haemophilia care in Europe. Haemophilia 2002;8: 33-43. Molho P, Rolland N, Lebrun T, Dirat G, Courpied JP, et al. Epidemiological survey of the orthopaedic status of severe haemophilia A and B patients in France. The French Study Group. Haemophilia 2000;6: 23-32. Aledort LM, Haschmeyer RH, Pettersson H. A longitudinal study of orthopaedic outcomes for severe factor-VIII-deficient haemophiliacs. The Orthopaedic Outcome Study Group. J Intern Med 1994;236: 391399. Pollmann H, Richter H, Ringkamp H, Jurgens H, Eur J Pediatr 1999;158 Suppl 3: S166-170. Ramgren O. Haemophilia in Sweden III Symptomatology, with special reference to differences between haemophilia A and B. Acta Med Scand 1962;171: 237-242 74 Prasant et al. / Indian J. Pharm. Biol. Res., 2014; 2(4):68-76 21. Rainsford SG, Hall A, A three-year study of adolescent boys suffering from haemophilia and allied disorders. Br J Haematol 1973;24: 539-551. 22. Blanchette P, Rivard G, Israels S, Robinson S, Ali K, et al. A survey of factor prophylaxis in the Canadian haemophiliaA population. Haemophilia 2004;10: 679683 23. Beltran-Miranda CP, Khan A, Jaloma-Cruz AR, Laffan MA, Thrombin generation and phenotypic correlation in haemophilia A. Haemophilia 2005;11: 326- 334 24. Shima M, Matsumoto T, Fukuda K, Kubota Y, Tanaka I, et al. The utility of activated partial thromboplastin time (aPTT) clot waveform analysis in the investigation of hemophilia A patients with very low levels of factor VIII activity (FVIII:C) ThrombHaemost 2002;87: 436441. 25. Arbini AA, Mannucci PM, Bauer KA, Low prevalence of the factor V Leiden mutation among "severe" hemophiliacs with a "milder" bleeding diathesis. ThrombHaemost 1995;74: 1255-1258. 26. EscuriolaEttingshausen C, Halimeh S, Kurnik K, Schobess R, Wermes C, et al. Symptomatic onset of severe hemophilia A in childhood is dependent on the presence of prothrombotic risk factors. ThrombHaemost 2001;85: 218-220. 27. Ghosh K, Shetty S, Mohanty D. Milder clinical presentation of haemophilia A with severe deficiency of factor VIII as measured by one-stage assay. Haemophilia 2001;7: 9-12. 28. Jayandharan GR, Nair SC, Poonnoose PM, Thomas R, John J, et al. Polymorphism in factor VII gene modifies phenotype of severe haemophilia. Haemophilia 2009;15: 1228-1236. 29. Shetty S, Vora S, Kulkarni B, Mota L, Vijapurkar M, et al. Contribution of natural anticoagulant and fibrinolytic factors in modulating the clinical severity of haemophilia patients. Br J Haematol 2007;138: 541544. 30. Handelsman JE. The knee joint in hemophilia. OrthopClin North Am 1979;10: 139-173. 31. Roosendaal G, Vianen ME, Wenting MJ, van Rinsum AC, van den Berg HM, et al. Iron deposits and catabolic properties of synovial tissue from patients with haemophilia. J Bone Joint Surg Br 1998;80: 540545. 32. Stein H, Duthie RB, The pathogenesis of chronic haemophilicarthropathy. J Bone Joint Surg Br 1981;63B: 601-609. 33. Hooiveld MJ, Roosendaal G, Jacobs KM, Vianen ME, van den Berg HM, et al. Initiation of degenerative joint damage by experimental bleeding combined with loading of the joint: a possible mechanism of hemophilic arthropathy. Arthritis Rheum 2004;50: 2024-2031. 34. Acharya SS, Kaplan RN, Macdonald D, Fabiyi OT, DiMichele D, et al. Neoangiogenesis contributes to the Review Article 35. 36. 37. 38. 39. 40. 41. 42. 43. 44. 45. 46. 47. 48. 49. 50. 51. development of hemophilic synovitis. Blood 2011;117: 2484-2493. Arnold WD, Hilgartner MW, Hemophilicarthropathy. Current concepts of pathogenesis and management. J Bone Joint Surg Am 1977;59: 287-305. van den Berg HM, De Groot PH, Fischer K, Phenotypic heterogeneity in severe hemophilia. J ThrombHaemost 2007;5 Suppl 1: 151-156. Handelsman JE, Glasser RA, Pathogenesis and treatment of hemophilic arthropathy and deep muscle hemorrhages. ProgClinBiol Res 1990;324: 199-206. Railton GT, Aronstam A, Early bleeding into upper limb muscles in severe haemophilia. Clinical features and treatment. J Bone Joint Surg Br 1987;69: 100-102. Gilbert MS, Musculoskeletal manifestations of hemophilia. Mt Sinai J Med 1977;44: 339-358. Ljung RC, Intracranial haemorrhage in haemophilia A and B. Br J Haematol 2008;140: 378-384. Chalmers E, Williams M, Brennand J, Liesner R, Collins P, et al. Guideline on the management of haemophilia in the fetus and neonate. Br J Haematol 2011;154: 208-215. Mahasandana C, Patharathienskul D, Suvatte V. Hemophilia with factor VIII and factor IX inhibitors, incidence, bleeding problems and management. Southeast Asian J Trop Med Public Health 1993;1: 106-112. Forbes CD BR, Prentice CR, Douglas AS, Gastrointestinal bleeding in haemophilia. Q J Med 1973;42: 503-511. Prentice CR, Lindsay RM, Barr RD, Forbes CD, Kennedy AC, et al. Renal complications in haemophilia and Christmas disease. Q J Med 1971;40: 47-61. Gitschier J WW, Goralka TM, Wion KL, Chen EY, Eaton DH, Vehar GA, Capon DJ, Lawn RM. Characterization of the human factor VIII gene. Nature 1984;312: 326-330 Vehar GA KB, Eaton D, Rodriguez H, O'Brien DP, Rotblat F, et al. Structure of human Factor VIII. Nature (London) 1984;312: 337-342. Pittman DD, Kaufman RJ, Proteolytic requirements for thrombin activation of anti-hemophilic factor (factor VIII).ProcNatlAcadSci U S A 1988;85: 2429-2433. Lenting PJ, van Mourik JA, Mertens K. The Life Cycle of Coagulation Factor VIII in View of Its Structure and Function. Blood 1998;92: 3983-3996. Lollar P, Parker ET. Structural basis for the decreased procoagulant activity of human factor VIII compared to the porcine homolog. J BiolChem 1991;266: 1248112486. Ananyeva NM, Kouiavskaia DV, Shima M, Saenko EL, Catabolism of the coagulation factor VIII: can we prolong lifetime of f VIII in circulation? Trends Cardiovasc Med 2001;11: 251-257. enting PJ, Neels JG, van den Berg BM, Clijsters PP, Meijerman DW, et al. The light chain of factor VIII comprises a binding site for low density lipoprotein 75 Prasant et al. / Indian J. Pharm. Biol. Res., 2014; 2(4):68-76 receptor-related protein. J BiolChem 1999;274: 2373423739. 52. Strauss H, The perpetuation of hemophilia by mutation. Pediatrics 1967;39: 186-193. 53. Vogel F, A probable sex difference in some mutation rates. Am J Hum Genet 1977;29: 312-319. 54. Naylor JA, Green PM, Rizza CR, Giannelli F, Analysis of factor VIII mRNA reveals defects in everyone of 28 haemophiliaA patients. Hum Mol Genet 1993;2: 11-17. 55. Lakich D, KazazianHH ,Antonarakis SE, Gitschier J, Inversions disrupting the factor VIII gene are a common cause of severe haemophilia A. Nat Genet 1993;5: 236-241. Cite this article as: Jiguru Prasant.Recent trends and advances in hemophilia – its management and new therapeutic outcomes. Indian J. Pharm. Biol. Res.2014; 2(4):68-76. All © 2014 are reserved by Indian Journal of Pharmaceutical and Biological Research This Journal is licensed under a Creative Commons Attribution-Non Commercial -Share Alike 3.0 Unported License. This article can be downloaded to ANDROID OS based mobile. Review Article 76