Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
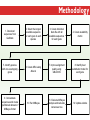
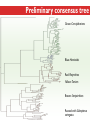
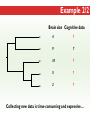
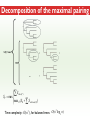
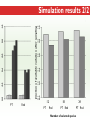
Three Weeks of Experience at the formatics Institute Christian Arnold Bioinformatics Group, University of Leipzig Bioinformatics Herbstseminar October 23th, 2009 Content 1. The 10kTrees Project 2. Phylogenetic Targeting 3. Acknowledgements 1.The 10kTrees Project Goals • Updated primate phylogeny that includes phylogenetic uncertainty – Use newest available sequence data, include as much primate species as possible, and update regularly – Produce a set of >=10,000 primate-wide trees (with branch lengths) that are appropriate for taxonomically broad comparative research on primate behavior, ecology and morphology using Bayesian methods • Make it accessible to other researchers Methodology 1. Download sequences from GenBank 2. Select the longest available sequence for each gene in each species 3. Create individual fasta file with all available sequences for each gene 4. Create availability matrix 5. Identify species with non-overlapping genes 6. Create MSA using Muscle 7. Improve alignment quality using GBLOCKS 8. Identify best substitution model for each gene 9. Concatenate sequences and create partitioned dataset in MrBayes format 10. Run MrBayes 11. Evaluate MrBayes analysis and calculate consensus tree 12. Update website Version 1 vs. Version 2 Version 1 Version 2 Species 187 231 Genes 4 mitochondrial (COI, COII, CYTB and ND1) and 1 autosomal gene (SRY) 6 mitochondrial (12S rRNA, 16S rRNA, COI, COII, CYTB, cluster of other mitochondrial genes) and 3 autosomal genes (SRY, CCR5, MC1R) Genetic loci 2 4 Total No. of Sites 5134 ~9000 Collected sequences 413 out of 935 total (55.8% missing data) 1007 out of 2079 total (51.6% missing data) No. of constraints 29 1 Generations 8 millions 60 millions Computing time ~ 48 days (16 processors in parallel, ~ 3 days each) ~ 2 years (32 processors in parallel, ~ 3 weeks each) Preliminary consensus tree Green: Cercopithecines Blue: Hominoids Red: Platyrrhines Yellow: Tarsiers Brown: Strepsirrhines Rooted with Galeopterus variegatus The 10kTrees Website http://10ktrees.fas.harvard.edu/ Current Progress • Submitted to Evolutionary Anthropology, in press. • Will be presented at the AAPA conference (April 2010) in Albuquerque, New Mexico • Version 2 is almost finished • Available at http://10kTrees.fas.harvard.edu Summary • Bayesian approach is time-consuming, but works well, even though data matrix is very sparse • Increased number of sequences in Version 2 dramatically reduces need for constraints and improves quality of tree and branch lengths estimates • Ongoing project • Total number of downloaded trees since June 2009: 95800 2. Phylogenetic Targeting Which species should we study? Goals For which species should we collect data in order to increase the size of comparative data sets ? Example 1/2 • Hypothesis: Two characters (x and y) show correlated evolution • Goal: Test this hypothesis comparatively (e.g. by using phylogenetically independent contrasts and correlation tests) • Problem 1: Data has been only collected for x, but not for y • Solution 1: Collect data for y and test hypothesis • Problem 2: From which species should we collect data for y? • Solution 2: Phylogenetic targeting!? Example 2/2 Brain size Cognitive data s1 4 ? s2 9 7 s3 10 ? s4 3 ? s5 2 ? Collecting new data is time-consuming and expensive… Methods • Systematically generate all possible pairwise comparisons • For every pairwise comparison, calculate character differences for the two species that form the pair and assign a score • Determine set of phylogenetically independent pairs that maximizes the sum of all selected pair scores (maximal pairing) s1 s2 s3 s4 s5 s6 s7 Maximal pairing: Example Decomposition of the maximal pairing S desc(T ) ST max max R ( S R S subtrees( R ) ) 2 Time complexity: O(n 3 ) , for balanced trees: O(n log 2 n) Simulation results 1/2 Detecting correlated character evolution, based on selection of 12 species • Random (Rnd) selection of species – Type 1 errors close to nominal level – Power: ~40%, independent of number of taxa – Uses 67% of available variation • Phylogenetic targeting (PT) induced selection of species – Type 1 errors close to nominal level – Power: 67-81%, increases with number of taxa – Uses 89% of available variation Fraction of available variation after sampling Simulation results 2/2 PT Rnd 12 PT 18 Rnd PT Rnd 24 PT Rnd Number of selected species Current Progress • A revised version will be resubmitted to American Naturalist in the not too distant future • TODO: Extend simulations and clarify some issues • Available at http://phylotargeting.fas.harvard.edu Summary • A focused selection of species can save valuable time and money • Phylogenetic targeting provides a very flexible approach and can address different questions in the context of limited resources • Dynamic programming algorithms are everywhere 3. Acknowledgements Thanks! • Harvard University • Max-Planck Institute for Evolutionary Anthropology • University of Leipzig • Charlie Nunn • Luke Matthews • Peter F. Stadler Any Questions? Thank you for your attention! Questions? If not: Cheers (it’s early, but not too early…)