Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
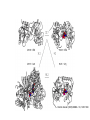
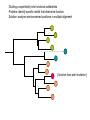
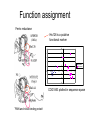
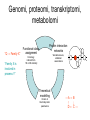
Proteiinianalyysi 52930 (3op) Liisa Holm Organisaatio • Luennot – 21.3.-18.4.2005, ke, pe 14-16, LS 1015 – kurssin kotisivu http://www.bioinfo.biocenter.helsinki.fi/downloads/teac hing/spring2006/proteiinianalyysi/ • Tentti – 25.4.2007, klo 14-16, LS 1015 • Oheislukemisto – Mount DW (2005) Bioinformatics. Sequence and genome analysis. 2nd edition. CSHL press, New York – Ch. 5-6,10-11 – Tramontano A (2005) The ten most wanted solutions in protein bioinformatics. Chapman & Hall/ CRC Mathematical Biology and Medicine series. Muut kurssit • Esitiedot: – Geneettinen bioinformatiikka 3 op • sekvenssivertailu • fylogeniapuut • Soveltaminen: – Proteiinianalyysin harjoitustyöt 5 op • webbityökalujen käyttö Johdanto Bioinformatics • An interdisciplinary science that synergistically utilizes the contribution of informatics, physics, and mathematics, but, ultimately, the objective is the solution of biological problems Protein bioinformatics • The goal is to assist experimental biology in assigning a function or suggesting functional hypotheses for all known proteins. Proteiinien merkitys • Proteiinit tekevät kaiken työn solussa ja ovat osallisina: – Geenisäätelyssä – Metaboliassa – Signaloinnissa – Tukirangassa – Kuljetuksessa – Solunjakautumisessa http://www.websters-online-dictionary.org/definition/english/ce/cell.html Proteins are a product of evolution • The basic principles of evolution must be kept in mind when new methods are devised or new routes are explored for inferring the function of a biological macromolecule. Proteiinit ovat erikoislaatuisia polymeerejä: • Tietyllä proteiinilla on aina sama aminohapposekvenssi – Proteiinin sekvenssi määräytyy DNAsekvenssin perusteella • Tietyllä proteiinilla on aina uniikki kolmiulotteinen rakenne. – Proteiinin rakenne määräytyy aminohapposekvenssin perusteella. aina = biologinen aina (poikkeuksia löytyy) Ei funktiota ilman rakennetta • Luonnon proteiinit laskostuvat spesifiseksi kolmiulotteiseksi rakenteeksi – komplementaarinen interaktiopartnerille • Denaturaatio tuhoaa funktion Evoluutio Sekvenssi – Rakenne - Funktio DNA-sekvenssi Luonnonvalinta Proteiinin sekvenssi Proteiinin funktio Proteiinin rakenne Evoluutioteoria Geenin kahdentuminen Perhe B Perhe A mutaatio mutaatio Perhe A’ mutaatio Koevoluutio A:B A B • Yhteinen kantamuoto => (jossain määrin) säilynyt rakenne ja funktio • Yhteinen valintapaine => säilyneet / muuntelevat alueet • Vertailu homologiin auttaa, kun hajautettu koodi on vaikea purkaa – DNA -> RNA -> Proteiini – Sekvenssi -> 3-ulotteinen rakenne -> Funktio – sukua informaatioteorialle Holm & Sander (1995) EMBO J 14, 1287-1293 Cell wall Many biosynthetic pathways GT3r (13) glycogen synthase GT35r (69) Glycogen Phosphorylase GT1i (467) UDP glucoronosyltransferases GT28i (50) b-NGlcNAc transferas e WecB (4) UDP-NGlcNAc 2epimerase GT4r (463) Sucrose and sucrosephosphate synthase GT5r (160) UDP-Glc glycogen and ADP-Glc starch glucosyltransferase UDP ADP PLP BG T (1) Energy storage Metabolic control point Cofactors: Phage T4 virulence factor The top ten ten most wanted solutions in protein bioinformatics 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. Protein sequence alignment Predicting protein features from sequence Function prediction Protein structure prediction Membrane proteins Functional site identification Protein-protein interaction Protein-small molecule interaction Protein design Protein engineering 1: Protein sequence alignment • Subproblems: homology detection alignment • Combined: maximize the probability of common ancestry (residue-residue) and of the existence of ancestor – Statistical significance tests – but, proteins are not randomly generated 1: Protein sequence alignment • Techniques: – scoring matrices – dynamic programming – Profiles – expectation maximization – Gibbs sampler – hidden Markov models 1: Protein sequence alignment • Functional continuity – Evolution-based inference • Duplication followed by divergence – Orthologues – same function – Paralogues – new function • Protein families – Multiple sequence alignment problem – Remote homology detection problem • Transitivity of homology Esimerkki sekvenssien evoluutiosta • Olettakaamme, että on olemassa meidän tuntemallemme rinnakkainen universumi, jossa kaikki proteiinit ovat neljän pituisia, aminohappoja on 26 (ABCDEFGHIJKLMNOPQRSTUVWXYZ) ja luonnonvalinta eliminoi kaikki sekvenssit, jotka eivät ole englanninkielessä esiintyviä sanoja. Toisin sanoen funktionaaliset proteiinit ovat nelikirjaimisia englannin sanoja. • Esitä muutama pistemutaatioiden ketju, joka muuttaa sekvenssin WORD sekvenssiksi GENE siten, että kaikki välivaiheet koodaavat funktionaalista proteiinia. Functional continuity WORD # WORE # GORE # GONE # GENE d=0 d=1 d=2 d=3 d=4 Montako funktionaalista sanaa on yhden mutaation päässä sanasta WORD? • WORD > • CORD, FORD, HORD, LORD, SORD, WARD, WOAD, WOED, WOLD, WORE, WORK, WORM, WORN, WORT Mikä osa kaikista mahdollisista neljän kirjaimen pituista merkkijonoista (funktionaalisuuteen katsomatta) on enintään etäisyydellä 1, 2, 3 tai 4 sanasta WORD? Etäisyydellä d on 4 Nd d sanaa, missä N on aakkoston koko. Yhteensä sanoja on N4. Osuus etäisyydellä 1 on 4/N3, etäisyydellä 2 osuus on 6/N2, etäisyydellä 3 osuus on 4/N ja etäisyydellä 4 se on 1. Sekvenssien (painotettukin) etäisyys on huono ”homologian” erittelijä, kun ei oteta huomioon positioden välisiä korrelaatioita. KVTMEHITT ## # RITMEHVTT ### RIHVLHVTT ## RIHVLHIST # RLHVLHIST # # LLHVLHLST # # RLHVAHLST # ## RLVVAHLGM # # RLVVDHLGK # # # DLVVDHMGR ## # LIVIDHMGR # ## KIVLAHMGR # ## KTVLAHMVH # KTVLAHGVH ### DDFLAHGVH # # # # ADRLGHGVR # # AERIGHGYR # # # TERLGHGYH Profile Superfamily Protein space 2: predicting features from sequence • Positive examples conservation problem • Positive/negative examples classification problem • Deterministic patterns • Probabilistic methods 2: predicting features from sequence • Functional fingerprints – E.g. Prosite patterns • • • • Secondary structure prediction Post-translational modification sites Sub-cellular localization Solvent accessibility • Techniques: regular expressions, profile, neural network Analog / homolog problem • Twilight zone in sequence similarity • Very broad twilight zone in structure similarity • Homologs share many features, including functional similarities Dividing a superfamily into functional subfamilies Problem: identify specific motifs that determine function Solution: analyse semiconserved positions in multiple alignment [ function-loss point mutation ] Function assignment Ferric reductase His126 is a putative functional marker 10 8 6 4 His absent His present 2 0 -8 -6 -4 -2 0 2 4 6 -2 -4 -6 COG1853 plotted in sequence space ‘FMN and nickel binding protein’ 3: Function prediction • Definition of “biological function” – Localization, process, biochemistry – Vocabulary • Protein names (synonyms) • Text mining • Function transfer – Error propagation • Intergration of transcriptomics and proteomics data – Gene co-expression, gene regulation – “Function = sum of interactions” Post-genomic view: Function = S interactions (From left to right, figures adapted from Olsen Group Docking Page at Scripps, Dyson NMR Group Web page at Scripps, and from Computational Chemistry Page at Cornell Theory Center). 4: Structure prediction • Energy calculations – Molecular mechanics, force field – Net stability of proteins ≈ 0 • Difficult to accurately model balance of entropic and enthalpic contributions • Searching conformational space – Energy minimization – Knowledge-based pseudo-potentials – Evolution-based (comparative modelling) levels of complexity in folding 4: Structure prediction • Levinthal’s paradox Polypeptidin rakenne • Polypeptidiketjun kolmiulotteisen rakenteen määrittävät torsiokulmat f ja y (oletetaan, että w=180 astetta). Kemiallisten sidosten rotaatiot suosivat energiaminimeitä. Esimerkiksi neliarvoinen hiili suosii gauche- (±60 astetta) ja trans- (180 astetta) orientaatioita. Näin ollen jokaisella aminohapolla on kymmenkunta mahdollista konformaatiota. • Montako mahdollista konformaatiota on N:n aminohapon pituisella proteiinilla? Jätä toistensa päälle osuvien atomien mahdollisuus huomiotta. – 10N • Kemiallisten sidosten tyypillinen rotaatiofrekvenssi on 1014 s-1. Kuinka kauan vähintään kestäisi, ennen kuin 40 aminohapon pituinen proteiini on käynyt läpi kaikki mahdolliset konformaationsa? – Hakuavararuus on 1040 konformaatiota, jaettuna 1014 s-1 tekee 1026 sekuntia. Vuorokaudessa on 86400 sekuntia ja vuodessa 31536000 sekuntia. Haku kestää ainakin 1018 vuotta. • Maailmankaikkeuden iäksi arvioidaan noin 10 miljardia vuotta. Montako proteiinia systemaattisella algoritmilla olisi ehtinyt laskostua peräkkäin aikojen alusta? Tulos tunnetaan Levinthalin paradoksina. • Luonnon proteiinit laskostumiseen kuluu aikaa millisekunneista minuutteihin. Luonnon laskostamisalgoritmi on 1029 ... 1024 kertaa tehokkaampi verrattuna systemaattiseen hakuun. 5: Membrane proteins • Special constraints due to lipid bilayer – Topography prediction • Three types of structures known – Porin beta-barrel – Helical bundles: bacteriorhodopsin, cytochrome oxidase, etc. – Light harvesting complex: full of chlorophyll 6: Functional site identification • Given a structure, can you tell what is the function? • Structural genomics – Targets families of unknown proteins • Techniques – 3D structure comparison + classification – Computational geometry Deduction Statistically significant sequence similarity Structure similarity Extant proteins Inferred ancestors 3D structure comparison has a longer look-back time Induction ? ? Properties of extant proteins ? Evolutionary continuity 7: Protein-protein interaction • Nature of interaction networks – “Scale-free” – Static / dynamic – Evolutionary robustness – Noisy data • Predicting interactions from sequence, from structure • Docking two structures into complex GroELcomplex Hemoglobin 1gr6 Molecular complexes via X-ray 30 S subunit of the ribosome Protein RNA 1fjg Hiivan proteiini-interaktioverkko Genomi, proteomi, transkriptomi, metabolomi “Q → Family X” “Family X is involved in process Y” Functional class assignment Protein interaction networks Wet-lab screens, statistical associations Homology inferred from 1D or 3D similarity Theoretical modelling Kinetic & thermodynamic parameters →A→B ↑ ↓ D←C→ 8: protein-small molecule interaction • Site identification • Ligand orientation • Prediction of affinity • Applications in pharmaceutical industry Enzymes • Catalytic triad: Asp, Ser, His 1CHO 9: Protein design • Design a sequence that will fold into a designated structure • “Paracelcus challenge” – Design a sequence more than 50 % identical to a natural protein such that the two proteins have different folds 10: Protein engineering • Modify native protein to endow it with new properties (not – yet – produced by natural evolution) Aminohappojen ominaisuuksia