Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
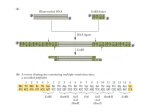
FUNGAL POPULATION LAB Overview: In this exercise, I’m going to ask you to take DNA sequence data, determine if collections from different geographical areas have sequence differences, identify restriction site differences and use these restriction site differences to tell me something about the population(s). Some Background: Eukaryotic Ribosomes are composed of two subunits and each subunit contains ribosomal RNAs and a number of proteins. The ribosomal RNAs (rRNAs) are transcribed together then processed to produce the separate rRNAs (Figure 1). The ribosomal RNA transcription block is repeated many times as a tandem repeat in the cell. All of the repeats are usually identical but the mechanism for “homogenization” is unknown.. Prokaryote rRNAs are smaller and do not exist as a tandem repeat. The eukaryotic 18S (Small ribosomal ) rRNA, 5.8S rRNA and ca. 27S (or large subunit ribosomal) RNAs are separated by transcribed spacers called internally transcribed spacer 1 (ITS1) and internally transcribed spacer 2 (ITS2). The region from the 3’ end of the 18S rRNA gene through the 5’ end of the LSU rRNA gene is variable at the species and sometimes population level and can be used to identify species, dissect species relationships and determine population structure. In this exercise, we will use this region to answer questions about the population structure of the common coral fungus Artomyces pyxidatus (correct names but most fungal books list it as Clavicorona pyxidata) 1 Exercise 1. Determine sequence differences. Open the file Fungallab.msf with any text editor including Word. [An msf file is an aligned sequence file produced by the Genetics Cooperative Group programs (GCG). GCG is available for our use on the University’s Unix computer “Larry” but you must have a Unix account to use it. These accounts are free to you as a student.] Print out a copy of the file. Examine the aligned sequences from different geographical regions and identify the differences in sequence that might be used to evaluate geographical structure. Mark these on your copy. Answer the following questions: Are there any differences between sequences from collections within the North and Central American collections? European and North American collections. What geographical regions show the most difference? Propose a hypothesis to suggest why this might be so. Yes, there are a few differences … around 160 and 440, 495. Between European and north American collection we have 101, 151, 500, 551, 601. The greatest difference seems to be between the European groups and the American groups (north and central). This could be because of historical patterns of dispersal. Exercise 2: Determine restriction site differences in North American collections Open the file Fungallab_fasta.rsf. This file presents the same data as the msf file but now each sequence is given separately and gaps in the sequence induced for alignment purposes have been removed. You will determine restriction sites for one European collection, one collection from Mexico and one collection from northeastern USA. Go to http://tools.neb.com/NEBcutter2/index.php This site contains a handy tool to map restriction sites for any given DNA sequence. Cut and paste the DNA sequence for one of the sequences in the Fungallab_fasta.rsf list. Select “all commercially available enzymes” and run the program. The program will display restriction sites for the entire sequence. You can mark a specific area of the sequence and zoom in on that area using options given on the results page. Locate regions in which there are differences between the North American collections and type in that section of DNA for one of the collections. Make sure to delete any gaps. Run the program and print the results for reference. Repeat this with the second collection. Answer the following questions: 2-1. What is a restriction enzyme? 2 An enzyme that cuts a DNA sequence of a certain code. 2-2. What restriction enzymes cut a sequence from Mexico/Costa Rica but don’t cut a sequence from eastern NA? BSAJ1 cuts mexico but not NA . QuickTime™ and a TIFF (Uncompressed) decompressor are needed to see this picture. QuickTime™ and a TIFF (Uncompressed) decompressor are needed to see this picture. 2-3. What restriction enzymes cut a sequence from Eastern North America but not Mexico/Costa Rica? 3 QuickTime™ and a TIFF (Uncompressed) decompressor are needed to see this picture. In using Taq1, we can cut the sequence of new york, TCGAGGTGAA, but there is no restriction enzyme for the maine cut, at TCAAGGTGAA, so we can differentiate between populations. Go to http://rebase.neb.com/rebase/rebhelp.html Search rebase for the enzymes you have identified as distinguishing between collections from different geographical regions. 2-4. What is the recognition sequence for each of the enzymes? Exercise 3: Determine the restriction fragment length polymorphisms (RFLPs) that you would see on a gel if you cut the ITS1-5.8S-ITS2 region with each enzyme you have identified. Go to: http://arbl.cvmbs.colostate.edu/molkit/mapper/index.html Copy and paste one of your ungapped sequences and click on “create map”. In the lower box, you will see a map by enzyme. Estimate fragment sizes on a gel from the data for each of the enzymes you have identified. Draw the patterns you would expect to see on a gel below. Repeat the process with your other sequence. 3-1. Can you identify the different sequences (haplotypes) by gel electrophoresis? How? Exercise 4: Mapping geographical patterns Open the file restriction pyx-haplotypes.rft in any text file editor including word. This file gives the restriction pattern for a number of collections. Map these collections on the map template attached. 4-1: Is there any geographical signal present or is this one large homogeneous population? 4-2: How do you explain discrepancies in the RFLP patterns such as 0,0 and 1,1 restriction patterns? What may be causing these? 4 4-3: If you consider North America as one large interbreeding population, is the population in Hardy Weinberg equilibrium? Calculate the haplotype frequencies and estimate the frequency of p2, 2pq, q2. How do these compare with observed frequencies. 4-4: What do you conclude about North American Artomyces pyxidatus Propose hypotheses that might explain your data. 5