Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
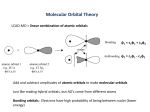
GENERAL CHEMISTRY 115 MO THEORY I. Shapes and Signs on Atomic Orbitals: Atomic orbitals are visual pictures of mathematical formulas, which, (when squared), tell us the probability of finding an electron at each possible position in space. These functions, like any mathematical function, have both positive and negative signs. For example, an s-type orbital is represented by a mathematical formula that is everywhere positive or everywhere negative. It makes no difference which, since the actual probability of finding the electron at a particular set of coordinates is given by the square of the orbital – i.e., the probability is always a positive value. The spherical shape of an s-type orbital tells us that the probability of finding the electron at a point in space depends only on how far the electron is from the nucleus, and not on the direction from the nucleus. However, p-type and d-type orbitals have non-spherical shapes. They have lobes which point at various different directions in space. Thus there will be a different probability for finding the electron in one direction than there will be in finding it in another direction. The s-type and p-type orbitals, with their accompanying signs, are shown below. Note that if we reverse ALL the signs in one of these orbitals, it will not change the probability distribution, since the probability will be given by squaring each picture. [And thus all negative lobes will become positive, once we square the wavefunction – i.e., the orbital.] + s-type + – px-type – + + – py-type pz-type In the pictures above, the three p-type orbitals are arbitrarily assigned as px, py, and pz. Each will always be aligned along whichever coordinate axis is given in its name – i.e., the px-type orbital is always aligned along the x-axis direction; the py-type orbital is always aligned along the y-axis direction; and the pz-type orbital is always aligned along the z-axis direction. Also, all three porbitals have identical forms – only their directions are different. [In the drawing above, the pz orbital’s lobes are coming in and out of the paper.] –1– Raynor: 6/27/2017 II. How to Combine Atomic Orbitals to Create Molecular Orbitals: Now, consider what happens when we bring two atoms together within a bond length of each other. Each atom brings with it all its atomic orbitals. The best overlap between them will occur when the lobes are aligned in the same directions, so we will bring together s-type with s-type; pxtype with px-type, etc. Also, we will be sure to apply the Conservation of Orbitals Rule. That is, if we mix together 2 atomic orbitals, we must get 2 new orbitals, which are called molecular orbitals. [In general, if we mix together n atomic orbitals, we must get n new molecular orbitals.] For the s-type orbitals, there are 4 different ways those orbitals can approach each other with their various signs: both can have + signs; both can have – signs; the left-one can be + and the right –; or the left one can be – and the right +. The possible overlaps and their resulting probabilities, (i.e., the square of the molecular orbital), are shown in the figure below. The left-most drawing shows the overlap of the atomic orbitals before any additions take place. The middle drawing shows the molecular orbital that results once we add together the two atomic orbitals. Finally, the right-hand drawing shows the square of the molecular orbital – i.e., this gives the probability of where an electron might be in space, if it occupies this particular molecular orbital. Figure 1: Molecular Orbitals Formed From the Overlap of s-type Orbitals 1 sA sB : + ++ + + 1 2 sA sB : – ––– – 2 3 sA sB : + +– – + – 3 4 sA sB : – –+ + – + 4 2 2 2 2 : + : + : + + : + + When we add together orbitals, we simply add the values from each function at each point in space. If the two values are both positive or both negative, then the magnitude will increase, with no change in sign. If one function has a positive value and the other a negative one at a particular point in space, then they will at least partially cancel each other, giving a result that is either smaller in magnitude or possibly 0, (if both have the same value with opposite signs). Thus the +/+ –2– Raynor: 6/27/2017 and –/– combinations both cause growth in the overlap region, i.e., the region along the bond axis, while the +/– and –/+ combinations both cause shrinkage. Since these are identical functions with opposite signs, in the +/– and –/+ cases a nodal plane is formed – this is a plane in space where the orbital, and thus the probability, is 0. This means there will be 0 probability of finding the electron at any point within the nodal plane. Since these atomic orbitals are identical, except for their signs, the nodal plane forms at the exact midpoint between the atoms, which is represented by dotted lines in Fig. 1. [A node is defined as a position in space where the wavefunction, and hence the probability distribution, is zero.] The nodal plane runs up-down and in-out in the +/– and –/+ drawings shown above. On the far right, in Fig. 1, is shown the probability distribution that is created by each molecular orbital – given by the square of the molecular orbital, in each case. Notice that wave functions 1 and 2 have identical probability distributions. This means that they represent the identical state for the system. Similarly, wave functions 3 and 4 have identical probability distributions and thus they also represent an identical state. This means that there are only 2 different states that can arise from the overlap of s-type orbitals – one from the +/+ combination of orbitals, (or the –/– combination), and one from the +/– combination of orbitals, (or the –/+ combination). We can thus eliminate the identical states and conclude that the overlap of 2 s-type orbitals yields 2 different molecular orbitals, (as expected from the Conservation of Orbitals rule). Let’s now compare the forms for the probabilities that arise for the +/+ and +/– combinations. When we focus on the bonding region, we see that the probability distribution arising from the +/+ combination predicts increased probabilities for finding an electron along the bonding region than would have occurred if no overlap had occurred. For this reason, we will refer to this molecular orbital as a bonding molecular orbital, (or MO). However, the probability distribution formed by the +/– combination predicts a decreased probability for finding an electron along the bonding region than would have occurred if there was no overlap. Since this means that the system would tend to break the bond, if this were the only occupied orbital, we will refer to this as an antibonding molecular orbital, (or MO). Note that electrons occupying bonding MOs will stabilize the bond in a molecule,) i.e., lower its energy), and electrons occupying anti-bonding MOs will destabilize it. For all of the p-type orbitals, we will repeat this process. I.e., we will create MOs from the +/+ and +/– linear combinations of each set of p-type atomic orbitals. The shapes of all the molecular orbitals that arise are shown in Table 1, below. [White shading means + signs and gray shading means – signs on each lobe. Dots are used to indicate the location of each nucleus in the homonuclear diatomic molecule.] Finally, we will label each MO according to its shape. Molecular orbitals that have lobes that include the bond axis are called -type MOs, (using the Greek letter sigma = ). Those that have oppositely-signed lobes on either side of the bond axis are called -type MOs, (using the Greek letter pi = ). If the MO is anti-bonding, we add a superscripted star, (*), to its label. If it is bonding in character, we add no star. We then identify the parent atomic orbitals by using their * names as subscripts on the MO. Thus the 2s MO is an anti-bonding sigma-type MO formed from overlapping the 2s orbitals on each parent atom and a 2 p MO is a bonding pi-type MO formed from the overlap of 2p orbitals on the parent atoms. The textbook uses the x, y and z designations –3– Raynor: 6/27/2017 on the labels from 2p orbitals. However, it uses a non-standard choice for its labels, so we will not do that. [The standard is to make the z-axis the same as the molecular axis, with the x-axis oriented up-down and the y-axis oriented in-out.] We will simply note whether the MO is from stype or p-type atomic orbitals and not worry about which p orbitals were involved. Since the shapes of the resulting MOs will be identical, regardless of the principle quantum number involved, we will get identical drawings as those shown in Fig. 1 for the overlap of any stype orbitals, i.e., those for 1s, 2s, 3s …, orbitals will all have the same overall shapes. Similarly, we will get identical shapes for all MOs from overlapping p-type atomic orbitals – regardless of their principle quantum numbers. Finally, note that the two bonding -type MOs are identical to one another, except for their orientations – one is oriented up-down and the other in-out. The same is true of the two antibonding *-type MOs. For this reason, both bonding -type MOs are identical in energy to one another and both anti-bonding *-type MOs are identical in energy to one another. –4– Raynor: 6/27/2017 Table 1: MOs from the 2s and 2p Orbitals Lin. Combo. AOs Before Mixing Molec. Orbital Irred. Rep. Type 2s(A)+2s(B) 2s Bonding 2s(A)–2s(B) 2s Antibonding 2pz(A)+2pz(B) 2 p Antibonding 2pz(A)–2pz(B) 2p Bonding 2px(A)+2px(B) 2p Bonding 2px(A)–2px(B) 2 p Antibonding 2py(A)+2py(B) 2p Bonding 2py(A)–2py(B) 2 p Antibonding –5– Raynor: 6/27/2017 III. Molecular Orbitals’ Energies: The exact order of the MO energies varies somewhat, depending on which atoms are bonded together. In all cases, the bonding and anti-bonding orbitals are grouped together by principle quantum number – i.e., all bonding and anti-bonding MOs from 1s-type orbitals are lower in energy than those from 2s and 2p orbitals, and all bonding and anti-bonding MOs from 2s and 2ptype orbitals are lower in energy than those from 3s, 3p and 3d orbitals, etc. For the atoms from Li2 through N2, the 2p orbitals are lower than the 2p orbital. However, for the atoms from O2 through Ne2, the order of these two levels is reversed. The relative orders for each case are shown in Figs. 2 and 3, where only the MOs from valence atomic orbitals are shown. [I.e., the 1s and 1s MO are not shown. They will both be filled with 2 electrons each and both lie below the levels arising from the 2s and 2p orbitals.] For elements in lower periods, the order will be the same, except that 3s and 3p orbitals will be involved for elements in the 3rd period; 4s and 4p orbitals for those in the 4th period, etc. Figure 2: Valence MO Order for Li2 through N2 2p* 2p* 2p* 2p 2p 2p 2p 2p 2s* 2s 2s 2s –6– Raynor: 6/27/2017 Figure 3: Valence MO Order for O2 through Ne2 2p* 2p* 2p* 2p 2p 2p 2p 2p 2s* 2s 2s 2s2s Note that in both Fig. 2 and Fig. 3 the atomic orbitals from the left atom appear on the left, those from the right atom appear on the right and the molecular orbitals, those obtained by mixing the atomic orbitals, are shown in the middle. To fill the MO levels with electrons, we use the same rules that we used when filling atomic orbitals. First, we use the aufbau principle. I.e., to create the lowest energy configuration, we start by placing electrons into the MO that is lowest in energy and able to accept more electrons. Then, once each energy level is filled, (with one spin up and one spin down within each sublevel), then, and only then, do we start to add electrons to the next level up in energy. If more than one level occurs that has the same energy, (as with the and * levels), we use Hund’s rule – i.e., we start by adding one electron with spin up to each sublevel, until all sublevels of the same energy have 1 electron each. Then, and only then, do we start to pair up electrons in those levels. For example, when adding electrons to the 2p levels, we first add 1 electron with spin up to each of the energyequivalent 2p levels. If more than 2 electrons are available, we then begin to add electrons with spin down to each of them. Note that we will only be adding the valence electrons to these levels. Recall that the number of valence electrons that an atom donates to the structure can be obtained from that atom’s group number: for atoms in groups 1-2, # valence e– = group #. For atoms in groups 13-18, # valence e– = group # – 10. For a neutral, (uncharged), diatomic molecule, the total number in the molecule will be twice the number for one atom. I.e., O2 will have 12 valence electrons, (= 26). If there is a charge on the system, then we need to account for it by subtracting electrons, if the charge is positive, or adding electrons, if the charge is negative. For example, O2–2 will have 14 valence electrons, which is 2 more than for the uncharged molecule. –7– Raynor: 6/27/2017 IV. MO Diagrams for the 2nd Period Homonuclear Diatomic Molecules: Figure 4 shows the occupancy of the MO levels for the homonuclear diatomic molecules Li2 through Ne2. Note the change in level order that occurs between N2 and O2. We can determine the bond order of the bond between the atoms, (i.e., whether the bond is a single bond, double bond, triple bond, etc.), as ½ times the net number of electrons that involve bonding. Since anti-bonding orbitals decrease the occupancy of electrons in bonding regions and bonding ones increase the occupancy in bonding regions, any electrons occupying anti-bonding MOs will negate the bonding effects of electrons in bonding MOs. This means that to determine the net number of electrons involved in bonding, we need to subtract the number of electrons occupying anti-bonding MOs from the number occupying bonding MOs. Thus, we get the following relationship for the bond order: 1 Bond Order = # e in bonding orbitals # e in antibonding orbitals . 2 A bond order of 1 indicates a single bond; of 2 indicates a double bond, etc. If the bond order is 0, then that tells us that no bond is expected – i.e., those atoms are not expected to bond together. This is the case when we calculate the bond orders for Be2 and Ne2, (as shown at the bottom of Fig. 4). Thus we do not expect Be or Ne to form stable homonuclear diatomic molecules, which is in agreement with experimental data. Any valence electrons that are in addition to the bonding electrons will be lone pair electrons – i.e., they will be localized on individual atoms, rather than shared in a bond. Thus, C2 is predicted to have a double bond. That means that 4 of its 8 electrons will be involved in bonds between the atoms. The remaining 4 electrons will occur in 2 lone pairs on the molecule – one pair on each carbon atom. Figure 4: MO Diagrams of the Second Period Homonuclear Diatomic Molecules 2p* 2p* 2p 2p 2s* 2p* 2p* 2p 2p 2s* 2s 2s Molecule: Li2 Be2 B2 C2 N2 O2 F2 Ne2 1 0 0 0 1 2 2 0 3 0 2 2 1 0 0 0 Bond Order #Unpaired e– –8– Raynor: 6/27/2017 Finally, some molecules have configurations with unpaired electrons in the molecule. This is the case with the ground-state configurations for B2 and O2. Note that for these molecules the highest energy electrons are the 2 electrons in -type MOs, one in each sublevel. Thus these electrons will remain as unpaired electrons in the molecule. When a molecule has one or more unpaired electrons, it is said to be paramagnetic. If all its electrons are paired, then the molecule is said to be diamagnetic. Paramagnetic molecules will behave differently from diamagnetic molecules in the presence of a magnetic field. If a stream of diamagnetic molecules passes through the poles of a magnet, nothing happens. However, if a stream of paramagnetic molecules passes through the poles of a magnet, half of the molecules will be attracted to one of the poles and the other half to the other pole. Thus the stream will break up into 2 separate streams. One of the more interesting cases is the case for O2. Note that the MO diagram for O2 predicts that it will have a double bond, (bond order = 2), and that it will be paramagnetic, (since it has 2 unpaired electrons). This is in perfect agreement with experimental data, which demonstrates the double bond-character of the bond in O2 and confirms that the molecule has 2 unpaired electrons. However, if we try to create a Lewis picture for O2, that picture cannot support both conclusions. Two possible Lewis structures for O2 are shown below. The left-hand picture is “perfect” – i.e., each O atom has an octet of 8 electrons around it and both atoms have formal charges of 0. It does predict a double bond for O2, but predicts that all electrons should be paired and thus O2 should be diamagnetic, in disagreement with experimental data. The right-hand picture is “imperfect” – each O atom has only 7 electrons in its vicinity. It does predict that O2 should have 2 unpaired electrons and thus be paramagnetic, but predicts only a single-bond between the two atoms, in disagreement with experimental data. O O O O Thus, neither Lewis picture is correct. In general, MO theory does a better job predicting the electronic structure of a molecule than does the overly simplistic Lewis diagram approach. Another interesting case is that for the C2 molecule. Curiously, all 4 of the electrons in its double bond are predicted to be -type electrons. With Valence Bond Theory, we found that double bonds consisted of one sigma-type and one pi-type bond. Recall that the sigma-type electrons have strong probabilities of occurring along the bond axis, but pi-type orbitals have 0 probabilities along the bond axis. Thus we normally expect that some of the electron density in the molecule will be directed along the bond axis. However, MO theory predicts that this will not happen in C2, since all its bonding electrons belong to pi-type orbitals and thus have 0 probability of existing along the bond axis. In C2, all of the electrons in -type MOs became non-bonding, since the ones in the bonding MOs were cancelled by electrons in anti-bonding MOs. These 4 electrons, therefore, become the lone pair electrons on each C atom. Thus C2 is predicted to have a double bond formed from 2 -type bonds with no -type bond. It is therefore not surprising that C2 is less stable than graphite or diamond, both of which have carbon atoms bound by -type bonds throughout their structures. B2 is another interesting molecule. It is predicted to have a single bond created entirely from electrons occupying -type MOs. But these electrons are unpaired! One occupies one of the bonding MOs and the other occupies the -bonding MO that is perpendicular to the first one. –9– Raynor: 6/27/2017