
HapMap PROJECT - Faculty of Science at Bilkent University
... • The correlation between SNPs is mediated by linkage disequilibrium (LD). – LD exists when alleles at distinctive loci occur together more frequently than expected given the known allele frequencies and recombination fraction between the loci. ...
... • The correlation between SNPs is mediated by linkage disequilibrium (LD). – LD exists when alleles at distinctive loci occur together more frequently than expected given the known allele frequencies and recombination fraction between the loci. ...


lecture26_Polymorphi..
... This analysis is based on 377 microsatellites in 1056 individuals from 52 populations. Variations within populations account for 93 to 95% of the data. Nonetheless we can identify clusters that are consistent with known populations. K is chosen in advance. For any given K, each individual is represe ...
... This analysis is based on 377 microsatellites in 1056 individuals from 52 populations. Variations within populations account for 93 to 95% of the data. Nonetheless we can identify clusters that are consistent with known populations. K is chosen in advance. For any given K, each individual is represe ...

The Genetic Basis of Idiopathic Pulmonary Arterial Hypertension
... gene may be linked to the development ofIP AH. BMPR2, a member of the transforming growth factor beta family, plays a key role in inhibiting cell growth and has been indirectly linked to cellular apoptosis. The aim of our study was to identify novel single nucleotide polymorphisms (SNPs) in the BMPR ...
... gene may be linked to the development ofIP AH. BMPR2, a member of the transforming growth factor beta family, plays a key role in inhibiting cell growth and has been indirectly linked to cellular apoptosis. The aim of our study was to identify novel single nucleotide polymorphisms (SNPs) in the BMPR ...

Structural Location of Disease-associated Single
... nsSNPs in shallow depressed or convex regions also cause disease - probably because these can also be binding pockets nsSNPs unlikely to be buried in protein – why? ...
... nsSNPs in shallow depressed or convex regions also cause disease - probably because these can also be binding pockets nsSNPs unlikely to be buried in protein – why? ...

A genome-wide association study of chronic otitis media with
... Objectives: Chronic otitis media with effusion (COME) and recurrent otitis media (ROM) have been shown to be heritable, but candidate gene and linkage studies to date have been equivocal. Our aim was to identify genetic susceptibility factors using a genome-wide association study (GWAS). Methods: We ...
... Objectives: Chronic otitis media with effusion (COME) and recurrent otitis media (ROM) have been shown to be heritable, but candidate gene and linkage studies to date have been equivocal. Our aim was to identify genetic susceptibility factors using a genome-wide association study (GWAS). Methods: We ...

Mining Single Nucleotide Polymorphisms from public sequence
... Software exists to mine them from public data sets, but this doesn’t work in real time. GRID technology could help to deliver up-to-date alignments to users for any query sequence with putative SNPs marked up. Related useful features would include bootstrapped trees for each alignment, generated on ...
... Software exists to mine them from public data sets, but this doesn’t work in real time. GRID technology could help to deliver up-to-date alignments to users for any query sequence with putative SNPs marked up. Related useful features would include bootstrapped trees for each alignment, generated on ...

Mining SNPs from public sequence Databases
... Software exists to mine them from public data sets, but this doesn’t work in real time. GRID technology could help to deliver up-to-date alignments to users for any query sequence with putative SNPs marked up. Related useful features would include bootstrapped trees for each alignment, generated on ...
... Software exists to mine them from public data sets, but this doesn’t work in real time. GRID technology could help to deliver up-to-date alignments to users for any query sequence with putative SNPs marked up. Related useful features would include bootstrapped trees for each alignment, generated on ...


Intro to Computational Genetics
... • If the number of tests is n, we set the threshold to be 0.05/n. • A very conservative test. If the tests are independent then it is reasonable to use it. If the tests are correlated this could be bad: – Example: If all SNPs are identical, then we lose a lot of power; the false positive rate reduce ...
... • If the number of tests is n, we set the threshold to be 0.05/n. • A very conservative test. If the tests are independent then it is reasonable to use it. If the tests are correlated this could be bad: – Example: If all SNPs are identical, then we lose a lot of power; the false positive rate reduce ...

Genome browser - Indiana University
... • Current data set – 1 SNP every 279 bp A much more complete variation resource by which the genome-wide map can evaluated ...
... • Current data set – 1 SNP every 279 bp A much more complete variation resource by which the genome-wide map can evaluated ...

SNPs
... • Investigate the genetic factors shared by the cases, but absent from the controls; i.e. find the associations between the genetic factors and the disease. • The most straightforward way: genotype all the individuals. • But this is far too expensive with current technology! ...
... • Investigate the genetic factors shared by the cases, but absent from the controls; i.e. find the associations between the genetic factors and the disease. • The most straightforward way: genotype all the individuals. • But this is far too expensive with current technology! ...


Trait Mapping - Nematode bioinformatics. Analysis tools and data
... How to use markers to find disease? genome-wide, dense SNP marker map ...
... How to use markers to find disease? genome-wide, dense SNP marker map ...

Figure 1, Multi-traits association study of WEIGHT, HIP, BMI and
... Factor analysis (FA) generally requires at least three variables to get a stable common factor. To compare with PC-based study, we conducted FA-based multivariate regression analysis of pleiotropic association in the first group comprised of WEIGHT, BMI, WAIST, and HIP. The results of significant SN ...
... Factor analysis (FA) generally requires at least three variables to get a stable common factor. To compare with PC-based study, we conducted FA-based multivariate regression analysis of pleiotropic association in the first group comprised of WEIGHT, BMI, WAIST, and HIP. The results of significant SN ...



genotyping single nucleotide polymorphisms located on
... We are designing primer extension assays to type SNPs located on the Y chromosome as well as in the mitochondrial genome in order to evaluate their usefulness in forensic applications. The results of these primer extension reactions are being analyzed using matrix assisted laser desorption-ionizatio ...
... We are designing primer extension assays to type SNPs located on the Y chromosome as well as in the mitochondrial genome in order to evaluate their usefulness in forensic applications. The results of these primer extension reactions are being analyzed using matrix assisted laser desorption-ionizatio ...
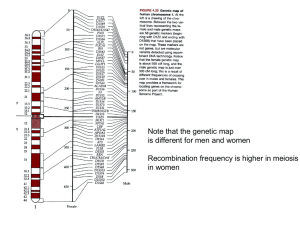
Lecture 3 Human Genetics
... Many human disorders, conditions and predispositions are multigenic Twin studies where identical twins are raised together or raised apart Look at complex behaviors and ask if they are genetic or environment Answer: For almost every single behavior…..it’s a little of both “Heritability” or the frac ...
... Many human disorders, conditions and predispositions are multigenic Twin studies where identical twins are raised together or raised apart Look at complex behaviors and ask if they are genetic or environment Answer: For almost every single behavior…..it’s a little of both “Heritability” or the frac ...

Analytical methods to identify genes for complex traits in Genome
... Current methods for GWA studies look for the association of simple DNA variants (eg, SNPs) with a complex trait of interest reducing the complexity of the approach to “n” simple univariate tests, with “n” equal to the total number of DNA variants under scrutiny. In this case, analyzing the genetic b ...
... Current methods for GWA studies look for the association of simple DNA variants (eg, SNPs) with a complex trait of interest reducing the complexity of the approach to “n” simple univariate tests, with “n” equal to the total number of DNA variants under scrutiny. In this case, analyzing the genetic b ...

A single-nucleotide polymorphism tagging set for human drug
... Carlson, C.S. et al. Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am. J. Hum. Genet. 74, 106−120 (2004). ...
... Carlson, C.S. et al. Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am. J. Hum. Genet. 74, 106−120 (2004). ...


No Slide Title
... – Mating is random (but there is stratification and assortative mating) – Population is large (if appropriately chosen!) – No mutation (but there is) – No migration (but migration occurs) – No selection (but there can be selection) ...
... – Mating is random (but there is stratification and assortative mating) – Population is large (if appropriately chosen!) – No mutation (but there is) – No migration (but migration occurs) – No selection (but there can be selection) ...
Tag SNP

A tag SNP is a representative single nucleotide polymorphism (SNP) in a region of the genome with high linkage disequilibrium that represents a group of SNPs called a haplotype. It is possible to identify genetic variation and association to phenotypes without genotyping every SNP in a chromosomal region. This reduces the expense and time of mapping genome areas associated with disease, since it eliminates the need to study every individual SNP. Tag SNPs are useful in whole-genome SNP association studies in which hundreds of thousands of SNPs across the entire genome are genotyped.