Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Crystallization wikipedia , lookup
Double layer forces wikipedia , lookup
Click chemistry wikipedia , lookup
Thermomechanical analysis wikipedia , lookup
Debye–Hückel equation wikipedia , lookup
Chemical potential wikipedia , lookup
Physical organic chemistry wikipedia , lookup
Electrochemistry wikipedia , lookup
Electrolysis of water wikipedia , lookup
Internal energy wikipedia , lookup
Chemical reaction wikipedia , lookup
Spinodal decomposition wikipedia , lookup
Marcus theory wikipedia , lookup
Ultraviolet–visible spectroscopy wikipedia , lookup
Photosynthetic reaction centre wikipedia , lookup
George S. Hammond wikipedia , lookup
Rate equation wikipedia , lookup
Bioorthogonal chemistry wikipedia , lookup
Stoichiometry wikipedia , lookup
Thermodynamics wikipedia , lookup
Stability constants of complexes wikipedia , lookup
Determination of equilibrium constants wikipedia , lookup
Chemical thermodynamics wikipedia , lookup
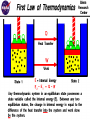
Chapter 2: Equilibrium Thermodynamics and Kinetics Equilibrium Thermodynamics: predicts the concentrations (or more precisely, activities) of various species and phases if a reaction reaches equilibrium. Kinetics tells us how fast, or if, the reaction will reach equilibrium. System: that portion of the universe we wish to study. It could be as simple as a beaker containing solution or as complex as the universe. A system can be open (exchanging matter and energy with its surroundings), closed (not exchanging matter with its surroundings), or isolated (exchanges neither matter nor energy with its surrounding.) Intensive properties: independent of the magnitude of the system (i.e. temperature and pressure.) Extensive properties: dependent on the magnitude of the system (i.e. volume and mass.) Phase: defined as a uniform, homogeneous physically distinct and mechanically separable portion of a system. Components: chemical constituents (species) needed to completely describe the chemical composition of every phase in a system. For example: the solid, liquid and vapor phases of H2O would be formed by combining the two components (H and O) in the proportions 2H + O → H2O. The First Law of Thermodynamics: (a.k.a. Conservation of Energy) Energy can neither be created nor destroyed, it can only be changed from one form to another. The change in internal energy of a system is equal to the heat added to the system minus the work done by the system. ΔE = q – w Where ΔE is the change in internal energy of the system, q is the heat added to or removed from the system, and w is the work done on or by the system. By convention, heat added to a system is positive and work done by a system is positive. Thus, the internal energy of a system will increase if heat is added and will decrease if work is done by the system. Internal energy: the energy associated with the random, disordered motion of molecules. Heat: energy in transit from a high temperature object to a lower temperature object. Work: forms of energy transfer which can be accounted for in terms of changes in the macroscopic physical variables of the system. ΔE = q – w What is the system? Where does the heat come from and what is the work being done? Is the heat being added to the system, or removed from the system? Is the work being done to or by the system? Remember: ΔE = q – w Now to the math… If the work done by or on a system causes a change in volume at constant pressure, then the equation for ΔE becomes: ΔE = q – PΔV. Enthalpy(H) is equal to the heat flow when processes occur at constant pressure and the only work done is pressurevolume work. Note: dH = dq at constant pressure. (see page 28 of text) Exothermic reactions: reactions that release heat energy (enthalpy is negative for the reaction). Reactants → products + heat CH4 + 2O2 → CO2 + 2H2O + heat Endothermic reactions: reactions that use heat energy (enthalpy is positive for the reaction). Reactants + heat → products Ba(OH)2·8H2O(s ) + 2NH4SCN(s ) → Ba(SCN)2(s ) + 10H2O(l ) + 2NH3(g ) Heat of formation: enthalpy change that occurs when a compound is formed from its elements at a specific (standard) temperature and pressure. The heat of formation for the most stable form of an element is arbitrarily set equal to zero. Second law of thermodynamics: for any spontaneous process, the process always proceeds in the direction of increasing disorder—entropy. Heat energy Another way to look at entropy is that during any spontaneous process, there is a decrease in the amount of usable energy. Consider the combustion of coal, an ordered complex organic molecule is through the process of combustion, broken down into ash, CO2, H2O, SOx & NOx. Resulting in a dramatic decrease in the amount of usable energy. Mathematically, entropy (S) is described in the following equation. DS = q/T Where DS is the change in entropy and T is the temperature in Kelvin. When the entropy equation is combined with the enthalpy equation and only pressure-volume work is considered, then: dH = TdS + VdP Systems can exist in several states: unstable, metastable and stable. Figure 2-1 in text A system at equilibrium is in a state of minimum energy. This energy can be measured either as: Helmholtz free energy when the reaction occurs at constant temperature and volume. Or Gibbs free energy when the reaction occurs at constant temperature and pressure. Let’s first concentrate on Gibbs free energy. For a system at constant T & P. Gibbs free energy can be written as: G = H – TS Where H is enthalpy(kJ mol-1), S is entropy(J mol-1K-1) and T is temperature (K) For changes that occur at constant T & P the expression for Gibbs free energy becomes: DG = DH – TDS If DG is (-), the process occurs spontaneously. If DG = 0, the process is at equilibrium If DG is (+), the reaction does not occur spontaneously To determine DGRº (for the entire reaction) you must subtract the SDG of the reactants from the SDG of the products. DGReaction = SDGProducts – SDGReactants See appendix II in the back of the book (page 474). So… DGR = DHR - TDSR Chemical potential mi = (DG / Dni) Where mi is the chemical potential of a certain component in a system and Dni is the change in moles of that component. For a system at equilibrium, mi is the same in all phases. The chemical potential, µ, of a component in a solution can be thought of in many ways: 1. A measure of the "escaping tendency" for a component in a solution; 2. A measure of the reactivity of a component in a solution; 3. The chemical potential of a component in a solution is defined as the rate at which the internal energy of the solution increases as the number of moles of the component in question increases, for a given entropy and volume of the solution. Activity and Fugacity (for a gas): the apparent (or effective) concentration of a species as opposed the the actual concentration. They are a measure of the departure of a system from ideal behavior and need to be taken into account even when dealing with relatively dilute solutions. Activity (or fugacity) is related to concentration through the activity coefficient (gi). gi = ai / mi Where ai is the activity and mi is the actual concentration. Incorporating the chemical potential into the activity yields the following equation: mi = moi + RT ln(ai) Where moi is the chemical potential of component i in its standard state. Now, a word or two about partial pressure… John Dalton studied the effect of gases in a mixture. He observed that the Total Pressure of a gas mixture was the sum of the Partial Pressure of each gas. P total = P1 + P2 + P3 + .......Pn The Partial Pressure is defined as the pressure of a single gas in the mixture as if that gas alone occupied the container. In other words, Dalton maintained that since there was an enormous amount of space between the gas molecules within the mixture that the gas molecules did not have any influence on the motion of other gas molecules, therefore the pressure of a gas sample would be the same whether it was the only gas in the container or if it were among other gases. So, if you ask "what is the fugacity, and how do I think about it", just think of it as a partial pressure: it is a strong function of the mole fraction of the component in the gas phase, and of the total pressure of the gas phase, just like a partial pressure; more precisely, remembering that chemical potential is a quantitative measure of the reactivity of a component in a phase, we can think of fugacity as a measure of how much the chemical potential of the component in the gas deviates from the chemical potential of some reference, namely, the standard state, due to changes in P and/or the mole fraction of the component i. Chemical Equilibrium Chemical equilibrium applies to reactions that can occur in both directions. In a reaction such as: CH4(g) + H2O(g) <--> CO(g) + 3H2(g) Many chemical reactions are reversible. The products formed react to give back the original reactants, even as the reactants are forming more products. After some time, both the forward and reverse reactions will be going on at the same rate. When this occurs, the reaction is said to have reached equilibrium. Equilibrium Constant To determine the amount of each compound that will be present at equilibrium you must know the equilibrium constant. To determine the equilibrium constant you must consider the generic equation: aA + bB <--> cC + dD The upper case letters are the molar concentrations of the reactants and products. The lower case letters are the coefficients that balance the equation. Use the following equation to determine the equilibrium constant (Kc also written Keq). For example, determining the equilibrium constant of the following equation can be accomplished by using the Kc equation. Using the following equation, calculate the equilibrium constant. N2(g) + 3H2(g) <--> 2NH3(g) A one-liter vessel contains 1.60 moles NH3, .800 moles N2, and 1.20 moles of H2. What is the equilibrium constant? 1.85 Incorporating Gibbs free energy with the equilibrium constant yields the following equation: logKeq = -DGR / 5.708 Where 5.708 is a combination of the gas constant (R) and standard temperature of 25°C. Example 2-1 Calculate the solubility product for gypsum at 25°C Note: [activity] and (concentration). Henry’s Law: The relationship between the equilibrium constant and the solubility product is used to describe the activity of a dilute component as a function of concentration. Where: ai = hiXi ai is the activity of species i, hi is the Henry’s law proportionality constant and Xi is the concentration of species i. Henry’s law relates the fugacity of a gas to its activity in solution. When a gas behaves ideally (1 atm and near surface temperatures) fugacity of a gas equals its partial pressure. Therefore, Henry’s law can be written: ci = KHPi Where ci is the concentration of the gaseous species i in solution, KH is Henry’s law constant in mol L-1 atm-1 (see table 2-1 pg 33), and Pi is the partial pressure of gaseous species i. Example 2-2 (page 33) Calculate the solubility of oxygen in water at 20°C. At sea level (a total atmospheric pressure of 1 bar; where one atm. = 1.0135 bar) the partial pressure of oxygen is 0.21bar. At 20°C, the Henry’s law constant for oxygen is 1.38 X 10-3 mol/L·bar. O2(aq) = (1.38 X 10-3 mol/L·bar)(0.21 bar) = 2.9 X 10-4 mol/L Or (2.9 X 10-4 mol/L)(32.0 gO2/mol) = 9.28 X 10-3 g/L or 9.28 mg/L Free energies at temperatures other than 25°C If the deviations in temperature from 25°C are small (~15°C or less) we can make the assumption that DHR and DSR are constant. This brings us to the van’t Hoff equation: lnKt = lnKr + (DHR/R)(1/Tr – 1/Tt) Where Kt is the equilibrium constant at temperature t, Kr is the equilibrium constant at 25°C, Tt is the temperature t and Tr is 298.15K. Of course, DHR is the enthalpy for the reaction, and R = 8.314 X 10-3 kJ/mol·K. Example 2-3 page 34. Calculate the solubility product of gypsum at 40°C using the van’t Hoff equation. For temperature changes greater than 15°C, computation becomes very complex and requires the use of a spread sheet or other computer program. To determine the correct enthalpy under these circumstances, heat capacity must be introduced into the calculation. Recall that heat capacity is the amount of heat energy required to raise the temperature of 1 gram of a substance 1°C. Cp = a + bT – c/T2 a,b & c are experimentally determined constants. (You may see cp which refers to heat capacity at a constant pressure, or cv which refers to heat capacity at a constant volume.) cp = a + bT – c/T2 With the heat capacity and several of the previous formulas, we can use the following two equations to determine the Gibbs free energy as a function of temperature (GT). HT = H298 + a(T-298) + b/2(T2 – 2982) + c(1/T – 1/298) ST =[a·ln(T/298) + b(T – 298) + c/2(1/T2 – 1/2982)] + S298 Le Chatelier's principle If a change is imposed on a system at equilibrium, the position of the equilibrium will shift in a direction that tends to reduce the change. http://www.mhhe.com/physsci/chemistry/essentialchemistry/flash/lechv17.swf To review: CaSO4·2H2O Ca2+ + SO42- + 2H2O + heat If there is an increase in Ca2+, what will happen? If the system’s temperature is decreased, what will happen? CaCO3 + SiO2 CaSiO3 + CO2(g) If the pressure on the system is increased, what will happen? Activity coefficient (g): measure of how a specific real system deviates from some reference system that is taken to be ideal. In an ideal solution, activity would equal concentration. The departure from ideal behavior is caused mainly by: •Electrostatic interactions between charged ions. •The formation of hydration shells around ions. There are a variety of models that are used to calculate activity coefficients. The next few slides will address these models without getting into the mathematical pathways. One factor that is important to understand is ionic strength (I). I = ½ Smizi2 Where mi = the moles/liter of ion i and zi is the charge of ion i. Debye-Hückel Model loggi = (-Azi2( I )1/2)/(1 + Bai( I )1/2) Where A and B are constants depending only on T and P and ai is the hydrated radius of a particular ion. Truesdell-Jones Model loggi = (-Azi2( I )1/2)/(1 + Bai( I )1/2) + bI Where b is an experimental constant. This equation includes the observation that high-ionic strength experimental systems the activity coefficients begin to increase with increasing ionic strength. Use Tables 2-2 and 2-3 for the necessary experimental constants. Example 2-4 Given the following river water chemistry, calculate the activity coefficient for Ca2+ at 25°C, using both the Debye-Hückel ands the Truesdell-Jones equations. Table 2-4 Appropriate Ranges of Ionic Strengths for Activity Coefficient Models Model Ionic Strength (mol/L) Debye-Hückel 0 to 0.1 Davies 0 to 0.6 Truesdell-Jones 0 to 2 Specific ion interaction 0 to 4 Pitzer 0 to 6 We should be aware of the different types of activity coefficient models and their limitations. Calculation of activity coefficients for uncharged species g = 100.1I Where I is ionic strength. This translates to: loggi = KiI Where Ki is a constant ranging in value from 0.02 to 0.23 at 25°C Environment Ionic Strength Activity coefficient estimate estimate Fresh water 0.002 1.00 Brackish water 0.02 1.00 Sea water 0.7 1.17 Activity of water When water is used as a solvent, pure liquid water at infinite dilution is used as the standard state; -- the activity of H2O = 1 at infinite dilution. The activity of water is related to the mole fraction of pure water, XH2O as follows: mH2O = moH2O + RTlnXH2O In most cases, we are dealing with dilute solutions and we can set the activity of H2O = 1. In more concentrated solutions, such as seawater, the activity will be slightly less than 1. An aqueous complex is a dissolved species formed from two or more simpler species, each of which can exist in aqueous solution. With the equation A+ + B- AB(aq) The equilibrium constant, in this case, is considered the stability constant (Kstab) because it is a measure of the stability of the aqueous complex. Kstab = [AB(aq)] / [A+][B-] [AB(aq)] = Kstab·[A+][B-] = Kstab·Ksp By setting the activity of the solid to 1, Ksp = [A+][B-] A Measure of Disequilibrium Consider the dissolution of gypsum: CaSO4·H2O Ca2+ + SO42- + 2H2O Ksp = [Ca2+][SO42-] This simplifies matters to the point that we can consider the activities of the products when determining Ksp. We would call this the activity product [AP] or ion activity product if only charged species are involved [IAP]. If the system is in equilibrium, the [AP] = Ksp. If the [AP] is less that the Ksp, the solution is undersaturated in gypsum. If the [AP] is greater than the Ksp, the solution is supersaturated with respect to gypsum. Example 2-6 Kinetics Equilibrium thermodynamics predicts the final state of a system. Kinetics tells us if the system will actually achieve this state within a reasonable time. It’s all relative… Homogeneous reactions: involve one phase (gas, liquid or solid. Heterogeneous reactions: involve two or more phases. Reactions often occur in a series of steps. The slowest step determines the rate at which the reaction will proceed. The dissolution of gypsum occurs in two main steps. 1st: energy input is required to free the ions from the crystal structure. 2nd: the ions must be diffused. If they are not, then the microenvironment surrounding the gypsum crystal becomes saturated and the reaction stops. CaSO4·H2O Ca2+ + SO42- + 2H2O Which ever these steps is the slowest, will determine the rate at which dissolution proceeds. A common environment for gypsum dissolution is in the presence of ground water. In this case, which step would determine the rate of dissolution? Order of reactions Zeroth order: The reaction rate is independent of the concentration of the reactant (A) t1/2 = 0.5A0/k First order: The reaction rate is dependent on the concentration of the reactant A B. t1/2 = 0.693/k Second order: The reaction rate is dependent on the concentration of the reactant 2A B. t1/2 = - 1/kA0 Note that the units for k depend upon the order of the reaction. See example 2-7 Comparison of 0th, 1st and 2nd orders Zero-Order Reaction For a zero-order reaction, the rate of reaction is a constant. When the limiting reactant is completely consumed, the reaction abruptly stops. Differential Rate Law: v=k The rate constant, k, has units of mole L-1 sec-1. First-Order Reaction For a first-order reaction, the rate of reaction is directly proportional to the concentration of one of the reactants. Differential Rate Law: v = k [A] The rate constant, k, has units of sec-1. Second-Order Reaction For a second-order reaction, the rate of reaction is directly proportional to the square of the concentration of one of the reactants. Differential Rate Law: v = k [A]2 The rate constant, k, has units of L mole-1 sec-1. Figure 2-4 Illustrates the variability of dissolution rates with change in pH. The Arrhenius Equation: relates the rate at which a reaction occurs to the temperature. k = A exp(-Ea/RT) Or log k = log A – (Ea/2.303RT) Where A is a experimentally derived factor, Ea is the activation energy for the reaction, R is the ideal gas constant and T is temperature in Kelvin. Activation energy: the energy that must be overcome in order for a chemical reaction to occur. *Eby notes that a rough rule of thumb for activation energy is that it doubles with every 10ºC increase in temperature. Nucleation Homogeneous nucleation occurs when a nucleus forms spontaneously in an oversaturated solution. Heterogeneous nucleation occurs when a nucleus forms in contact with a, usually solid, surface. The free energy of formation of a nucleus consists of the energy gained from the formation of bonds and the energy required to create the surface. DGnuc = DGbulk + DGsurf For an oversaturated solution, DGbulk is always negative. DGbulk can also be written: DGbulk = (4pr3/3V)kBT ln(a/ao) Where (4pr3/3V) is the volume of a spherical nucleus, V is the molecular volume, kB is the Boltzman constant (1.3805 X 10-23 J/K), T is the temperature in Kelvin, a is actual activity and ao is the activity for a saturated solution. DGsurf can also be written: DGsurf = 4pr2g Where g is the interfacial energy. This yields the final equation of: DGnuc = - (4pr3/3V)kBT ln(a/ao) + 4pr2g Finally….. The rate at which nuclei form can be determined from the standard Arrhenius rate equation. Rate of nucleation = A exp(-DG*/kBT) Where A is a factor related to the efficiency of collisions of ions or molecules, DG* is the maximum energy barrier, kB is the Boltzmann constant and T is the temperature in Kelvin.


























































![[A, 8-9]](http://s1.studyres.com/store/data/006655537_1-7e8069f13791f08c2f696cc5adb95462-150x150.png)








