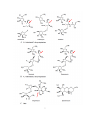
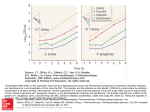
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
THE INVOLEVEMENT OF A PUTATIVE TWO COMPONENT SYSTEM PA2797-PA2798 IN PSEUDOMONAS AERUGINOSA AMINOGLYCOSIDE RESISTANCE by Raya Assan A thesis submitted to the Department of Biomedical & Molecular Sciences In conformity with the requirements for The degree of Master of Science Queen’s University Kingston, Ontario, Canada (August, 2014) Copyright ©Raya Assan, 2014 Abstract Pseudomonas aeruginosa is an opportunistic pathogen, being able to live in a diverse environmental niche. It possesses several resistance mechanisms, including a highly impermeable outer membrane and multiple RND-family efflux pumps, against a variety of antimicrobials, including aminoglycosides. P. aeruginosa causes a variety of infections in individuals, especially those with compromised immune system. Chronic lung infection in cystic fibrosis (CF) patients is an example of such. The most common antibiotic used today to treat the P. aeruginosa pulmonary infection in CF patients belongs to one of the major antibiotic families, aminoglycosides. Due to constant exposure of P. aeruginosa to aminoglycosides, this bacterium has gradually developed high level of resistance to aminoglycosides through the acquisition and selection of numerous chromosomal mutations. Previous P. aeruginosa transposon mutant library screen identified genes contributing to aminoglycoside resistance, including an atypical regulatory system, PA2797-PA2798. The array study on P. aeruginosa K767 ΔPA2798 determined possible pathways by which PA2797-PA2798 was connected to aminoglycoside resistance. A series of in-frame gene deletion (anr), gene cloning (rpoS, anr, rpsP operon), double deletion (rpoS and anr in K767 ΔPA2798), q-RT PCR (pslA, lexA, recN, rpsP operon), and a growth assay (K767, K767 ΔPA2797, K767 ΔPA2798) was performed to examine a possible connection between PA2797-PA2798, and RpoS, Anr, oxidative stress response, temperature stress response, and ribosomal proteins. Although the aforementioned experiments were performed successfully, none of them could identify a potential pathway through which PA2797-PA2798 was connected to aminoglycoside resistance. ii Co-Authorship I would like to acknowledge the contribution of Thomas L. Krahn to this work since he was responsible for the deletion of genes PA2797, PA2798, and rpoS in P. aeruginosa K767. iii Acknowledgements I would like to thank my supervisor, Dr. Keith Poole. This work could have not been done without his continuous help and support. Thank you for your patience, understanding, and guidance you have given me during these two wonderful years I spent as a member of your lab. I was truly blessed having you as my supervisor. Special thanks to Dr. Calvin Lau, for his support and friendship; you are such an amazing person. Thanks to wonderful Jerry Denning for his moral support; you are a true friend. I would like to thank Dr. Kenneth Jarrell, Dr. Bruce Banfield, and Dr. Nancy Martin for their guidance. I would also like to thank all those wonderful people I’ve worked with. Christie, you’re such an inspiration; Mike, Tom, Thomas, Andrew, thanks for putting up with my never ending list of questions. Alaya and Erin you girls rock! Thank you Sakeesnan, Daniel, and Hossam; I enjoyed every moment I’ve shared with you. At last, I would like to thank my parents for always being by my side, no matter how tough life gets. You’re my best friends from cradle to grave. iv Table of Contents Abstract ........................................................................................................................................... ii Co-Authorship................................................................................................................................ iii Acknowledgements ........................................................................................................................ iv List of Figures ............................................................................................................................... vii List of Tables ............................................................................................................................... viii List of Abbreviations ..................................................................................................................... ix Introduction................................................................................................................. 1 1.1 Pseudomonas aeruginosa...................................................................................................... 1 1.2 Aminoglycoside antibiotics ................................................................................................... 3 1.2.1 Aminoglycosides chemical structure and properties ...................................................... 4 1.2.2 Aminoglycosides uptake and mechanism of action ....................................................... 4 1.2.3 Aminoglycosides resistance mechanisms in P. aeruginosa ......................................... 16 1.2.3.1 Aminoglycoside modifying enzymes .................................................................... 16 1.2.3.2 Target modification ................................................................................................ 22 1.2.3.3 Impermeability resistance ...................................................................................... 23 1.2.3.4 Biofilm ................................................................................................................... 26 1.2.3.5 Mutational resistance ............................................................................................. 28 1.3 Statement of purpose ........................................................................................................... 30 Materials and Methods............................................................................................. 34 2.1 Bacterial strains and growth conditions .............................................................................. 34 2.2 DNA protocols .................................................................................................................... 34 2.3 Construction of genetic deletion in P. aeruginosa .............................................................. 38 v 2.3.1 Deletion of anr in P. aeruginosa K767 ........................................................................ 42 2.3.2 Deletion of PA2798 in P. aeruginosa .......................................................................... 42 2.4 Assessment of antimicrobial susceptibility ......................................................................... 43 2.5 Growth assay ....................................................................................................................... 43 2.6 Gene cloning ....................................................................................................................... 43 2.6.1 Cloning anr ................................................................................................................... 44 2.6.2 Cloning rpoS ................................................................................................................. 44 2.6.3 Cloning rpsP operon ..................................................................................................... 45 2.7 Quantitative real-time PCR ................................................................................................. 45 Results ........................................................................................................................ 47 3.1 The connection between RpoS and P. aeruginosa aminoglycoside resistance................... 47 3.2 The connection between Anr and P. aeruginosa aminoglycoside resistance ..................... 55 3.3 The connection between oxidative stress and P. aeruginosa aminoglycoside resistance ... 59 3.4 The connection between temperature stress and P. aeruginosa aminoglycoside resistance ................................................................................................................................................... 62 3.5 The connection between ribosomal genes and P. aeruginosa aminoglycoside resistance . 65 Discussion .................................................................................................................. 71 Conclusion ................................................................................................................. 81 References .................................................................................................................................... 83 vi List of Figures Figure 1.1 Amino-cyclitol ring of aminoglycoside antibiotics. ..................................................... 5 Figure 1.2 Aminoglycoside structure and classification. ............................................................... 6 Figure 1.3 Aminoglycoside uptake and mode of action. ............................................................... 9 Figure 1.4 Ribosome sites of translation. ..................................................................................... 12 Figure 1.5 P. aeruginosa aminoglycoside modifying enzymes. .................................................. 18 Figure 3.1 Impact of loss of rpoS on the expression of pslA. ...................................................... 50 Figure 3.2 Impact of loss of PA2798 on norB expression in P. aeruginosa. .............................. 58 Figure 3.3 Impact of loss of PA2798 on lexA and recN expression in P. aeruginosa. ................ 63 Figure 3.4 Impact of loss of PA2797-PA2798 on growth of P. aeruginosa................................ 66 Figure 3.5 Impact of loss of PA2798 on rpsP expression in P. aeruginosa K767. ..................... 67 Figure 3.6 Expression of rpsP as a measure of rpsP operon expression in P. aeruginosa K767 ΔPA2798. ...................................................................................................................................... 70 Figure 4.1 The regulation of σF and σB in B. subtilis. .................................................................. 73 vii List of Tables Table 1.1 P. aeruginosa aminoglycoside modifying enzymes. ................................................... 19 Table 1.2 Genes whose disruption by a transposon insertion decreased aminoglycoside resistance ....................................................................................................................................... 32 Table 1.3 Genes whose expression was changed in K767 ΔPA2798 and were investigated in this study .............................................................................................................................................. 33 Table 2.1 Bacterial strains used in this study ............................................................................... 35 Table 2.2 Plasmids used in this study .......................................................................................... 36 Table 2.3 Primers used in gene deletion (5’ to 3’ order).............................................................. 39 Table 2.4 Primers used in gene cloning (5’ to 3’ order) .............................................................. 40 Table 2.5 Primers used in q-RT PCR (5’ to 3’ order) .................................................................. 41 Table 3.1 Impact of loss of rpoS on aminoglycoside resistance in P. aeruginosa....................... 49 Table 3.2 Impact of cloned rpoS on aminoglycoside resistance in K767 .................................... 52 Table 3.3 Impact of cloned rpoS on aminoglycoside resistance in K767 ΔPA2797 ................... 53 Table 3.4 Impact of cloned rpoS on aminoglycoside resistance in K767 ΔPA2798 ................... 54 Table 3.5 Impact of loss of rpoS on aminoglycoside resistance in K767 ΔPA2798 ................... 56 Table 3.6 Impact of loss of anr on aminoglycoside resistance in K767 ΔPA2798 ..................... 60 Table 3.7 Impact of cloned anr on aminoglycoside resistance in K767 ...................................... 61 Table 3.8 Impact of cloned rpsP operon on aminoglycoside resistance in K767 ΔPA2798 ....... 69 viii List of Abbreviations AAC aminoglycoside N-acetyltransferase Acetyl-CoA Acetyl coenzyme A ADP Adenosine-5’-diphosphate AME aminoglycoside modifying enzyme AMI amikacin AMP adenosine-5’-monophosphate ANT aminoglycoside O-nucleotidyltransferase APH aminoglycoside O-phosphotransferase ATP adenosine-5’-triphosphate c-di-GMP bis-(3’-5’)-cyclic di-guanosine monophosphate CAM chloramphenicol CAR carbenicillin cDNA complementary deoxyribonucleic acid CF cystic fibrosis CM cytoplasmic membrane DMSO dimethyl sulfoxide DNA deoxyribonucleic acid dNTP deoxyribonucleoside triphosphate EDPI energy-dependent phase I EDPII energy-dependent phase II GEN gentamicin HIV human immunodeficiency virus KAN kanamycin KDO 3-deoxy-D-manno-oct-2-ulosonic acid L-agar L-broth with 1.5% (wt/vol) agar L-broth Luria broth with 2.5g NaCl added LPS lipopolysaccharide MDR multidrug resistance ix MIC minimum inhibitory concentration NADH nicotinamide adenine dinucleotide OM outer membrane PAR paromomycin PCR polymerase chain reaction q-RT PCR quantitative real-time polymerase chain reaction ROS reactive oxygen species RND resistance nodulation division rRNA ribosomal ribonucleic acid tRNA transfer ribonucleic acid SPC spectinomycin STR streptomycin TET tetracycline TOB tobramycin TP thermopol buffer x Introduction 1.1 Pseudomonas aeruginosa Pseudomonas aeruginosa is a Gram-negative human pathogen, found in a multitude of environments, including water, and most commonly soil (Silby et al., 2011; Vasil, 1986). It is considered to be strictly aerobic, and not being able to ferment, but due to its genetic adaptability it can also live in anoxic environments through denitrification, where the alternative electron acceptors are nitrite, nitrate, and nitrogen oxide (Stanier et al., 1966; Trunk et al., 2010). As an opportunistic human pathogen (Vasil, 1986), P. aeruginosa rarely colonizes healthy individuals, typically causing a variety of community-acquired (Vasil, 1986) as well as nosocomial infections in immunocompromised patients (Floret et al., 2009; Strateva and Yordanov, 2009), including those with burn, cancer and HIV, where it causes urinary tract infection, chronic respiratory disease, pneumonia, bacteremia, and sepsis among many other complications (Driscoll et al., 2007; Almagro et al., 2012; Schuster and Norris, 1994; Gudiol et al., 2011; Pednekar et al., 2010). Compared to other bacterial pathogens, P. aeruginosa nosocomial infections are associated with high morbidity and mortality (Lister et al., 2009; Osmon et al., 2004). It also demonstrates a high incidence of infection in cystic fibrosis (CF) patients (Burns et al., 2001; Emerson et al., 2002). Diffuse pan-bronchiolitis and respiratory disease in individuals suffering from CF are two common chronic respiratory infections caused by P. aeruginosa (Lyczak et al., 2002). Treatment of these respiratory infections in CF patients is often made difficult due to P. aeruginosa innate antimicrobial resistance and/or the development of resistance, limiting the choice of effective treatments (Bonomo and Szabo, 2006). More 1 problematic, P. aeruginosa often demonstrates a multidrug resistance (MDR) phenotype (Foweraker, 2009). It is known to be resistant to a variety of antibiotics, including the three major bactericidal classes of antibiotics, β-lactams (Alvarez-Ortega et al., 2011), fluoroquinolones (Dotsch et al., 2009; Breidenstein et al., 2008; Gallagher et al., 2011; Schurek et al., 2008) and aminoglycosides, used to treat chronic P. aeruginosa infections in the lungs of CF patients (Livermore, 2002; Wang et al., 2006). Antibiotic-resistant P. aeruginosa infections have been linked to increases in morbidity and mortality, time spent in hospitals, need for surgical intervention, and cost of care (Lister et al., 2009). Resistance mechanisms against antibiotics are typically classified as intrinsic, adaptive, and acquired (Fernández et al., 2011). Intrinsic resistance mechanisms are chromosomally encoded, and constitutively expressed. Adaptive resistance mechanisms are also chromosomally encoded but are induced by environmental conditions. Acquired resistance mechanism results from acquisition of exogenous genes, typically on plasmids and transposons (Lister et al., 2009; Poole, 2011). Intrinsic resistance to antibiotics in P. aeruginosa is due to the existence of several properties (Breidenstein et al., 2011), (explained in more details in section 1.2.3), including a highly impermeable outer membrane (Yoshimura and Nikaido, 1982), the presence of several chromosomally-encoded multidrug efflux pumps (Jana and Deb, 2006; Magnet and Blanchard, 2005), the formation of biofilms (Lopez et al., 2010), and the presence of a small sub-population of microbial cells termed persisters (Lewis, 2008). The last three resistance mechanisms (efflux pumps, biofilm, and persisters) fall in to adaptive class of resistance as well. Modifying the structure of antimicrobials using inactivation enzymes (Ramirez and Tolmasky, 2010), as well as modifying the structure of antimicrobials’ target sites by methylation (Doi and Arakawa, 2007), are examples of the acquired resistance mechanisms present in P. aeruginosa. The presence of a 2 variety of resistance mechanisms in P. aeruginosa, from intrinsic resistance mechanisms to resistance mechanisms adopted from horizontally-transferred plasmids and chromosomally integrated DNA, enhances P. aeruginosa antimicrobial resistance and compromises treatment of P. aeruginosa infections (Mesaros et al., 2007). All of the aforementioned resistance mechanisms are important in resisting the activity of one of the major classes of antibiotics, aminoglycosides (Poole, 2011; Vakulenko and Mobashery, 2003; Jana and Deb, 2006). Aminoglycosides are important therapeutic agents that are commonly used to treat P. aeruginosa infections (Cheer et al., 2003; Durante-Mangoni et al., 2009; Jana and Deb, 2006). In recent years however, P. aeruginosa resistance mechanisms against this antimicrobial have become more developed (MacLeod et al., 2000; Shawar et al., 1999). Therefore, identifying factors involved in P. aeruginosa resistance to aminoglycoside has been recently a major part of investigation. 1.2 Aminoglycoside antibiotics Antimicrobial agents inhibit or disrupt essential cellular processes. They can be bacteriostatic, meaning they inhibit bacterial growth, or bactericidal, meaning they produce a killing event. Aminoglycosides are natural (neomycin, kanamycin, tobramycin, gentamicin, and paromomycin) as well as synthetic (amikacin, netilmicin, dibekacin, arbekacin, and isepamicin) bactericidal antibiotics (Shakil et al., 2008). Aminoglycoside bactericidal activity is mainly due their protein synthesis inhibition property. Aminoglycoside molecules bind to the bacterial 30S ribosomal subunit and perturb the translation process, which results in the production of aberrant proteins, and finally cell death (McCoy et al., 2011). Aminoglycoside uptake is dependent on the energy provided by the respiratory pathway and electron transport chain. As such, they are more effective against aerobic bacteria (Bryan et al., 1979; Bryan and Kwan, 1983). 3 1.2.1 Aminoglycosides chemical structure and properties The structure of most aminoglycosides consists of a backbone of an amino-cyclitol ring, which is a cycloalkane saturated with hydroxyl and amine groups, linked to amino-sugars (Silva and Carvalho, 2007). The majority of clinically used aminoglycosides, are typically 2deoxystreptamine (Figure 1.1) (Jana and Deb, 2006), although streptomycin possesses a streptidine molecule instead (Figure 1.1). Based on the positioning of the amino-sugars on the cycloalkane backbone, aminoglycosides are structurally divided into three types (Jana and Deb, 2006) (Figure 1.2). These three types are distinguished based on whether the amino-sugars bind to the 4, 6 position of the central cycloalkane, or the 4, 5 position, or neither. Aminoglycosides are positively-charged compounds (Silva and Carvalho, 2007) that are highly soluble in water and relatively insoluble in lipids. Due to their cationic state, they are able to bind to the negatively-charged molecules in the cells, such as DNA, RNA, and phospholipids. Aminoglycosides are also able to bind to the negatively-charged lipopolysaccharide of the bacterial cell wall (Jana and Deb, 2006); this is crucial to their uptake by the outer membrane. Although aminoglycosides are the most common antimicrobial agents used to treat P. aeruginosa infections, there is a negative side to their consumption. These antimicrobials have negative impacts on a patient’s organs, such as nephrotoxicity (poisonous effect on the kidneys), and ototoxicity (poisonous effect on the ears) (Silva and Carvalho, 2007). Although aminoglycoside use has toxic side effects, failure to develop new antibiotics with the same or better efficacy has promoted research on how to minimize aminoglycoside side effects. 1.2.2 Aminoglycosides uptake and mechanism of action Aminoglycoside antibiotics penetrate aerobically growing bacteria in three independent stages; an energy-independent step (ionic binding), energy-dependent phase I (EDPI) 4 Figure 01.1 Amino-cyclitol ring of aminoglycoside antibiotics. The amino-cyclitol ring is a cycloalkane saturated with hydroxyl and amine groups. In the majority of clinically used aminoglycosides, the cycloalkane is either 2-deoxystreptamine (most aminoglycosides) or a streptidine (streptomycin). The figure is taken from Jana and Deb (2006). 5 Figure 01.2 Aminoglycoside structure and classification. Figure is modified from the original figure by Jana and Deb (2006). (A) 4, 6-disubstituted 2-deoxystreptamines. Amino-sugars are attached to streptamine at positions 4 and 6. Most of the clinically useful aminoglycosides, such as gentamicin and tobramycin possess this structure. The arrows show the streptamine core. (B) 4, 5-disubstituted 2-deoxystreptamines. Amino-sugars are attached to streptamine at positions 4 and 5. Aminoglycosides, such as neomycin and paromomycin possess this type of the structure. The arrows show the streptamine core. (C) Non-standard aminoglycosides, including spectinomycin and streptomycin. In streptomycin, the amino-sugar is attached to a core known as streptidine at a single position. Spectinomycin lacks an amino-sugar in its structure. The arrow shows the streptidine core. 6 4 4 4 6 6 6 4 4 5 5 7 energy-dependent phase II (EDPII) (Bryan and Van Den Elzen, 1977) (Figure 1.3). In the ionic binding phase, positively-charged aminoglycosides bind to the negatively-charged part of the lipopolysaccharide (LPS), the core oligosaccharide (the phosphate component along with the sugar component, also known as KDO) (Taber et al., 1987). It appears that by binding to the negative part, aminoglycosides displace Mg2+ and Ca2+ ions that function as cross-bridging and link the adjacent lipopolysaccharide molecules. This displacement, disrupts the cross-bridging of LPS molecules, damages the outer membrane and enhances its permeability (Hancock et al., 1981; Loh et al., 1984). The outer membrane perturbation allows for the diffusion of the aminoglycosides in to the periplasm (Hancock et al., 1981; Martin and Beveridge, 1986) (Figure 1.3 step 2). The ionic binding phase is completely energy independent and uptake of aminoglycosides across the outer membrane is dependent on aminoglycoside concentration gradient (Taber et al., 1987). Stages two and three, EDPI and EDP II, transport aminoglycosides across the cytoplasmic membrane (CM). These two steps are energy-dependent. The electrochemical potential across the cytoplasm as well as the electron flow through the membrane bound respiratory chain provide the energy needed for both steps (Taber et al., 1987). EDPI is the initial step of aminoglycoside uptake from the periplasm to the cytoplasm. In EDPI, initial small amount of aminoglycosides traverse the CM (Figure 1.3 step 3). The dependence of aminoglycoside uptake on EDPI on a functional respiratory chain explains intrinsic resistance of anaerobes to aminoglycosides. In contrast with aerobic respiration, anaerobic respiration does not release sufficient energy for EDPI stage to occur (Bryan and Kwan, 1983; Bryan et al., 1979). In E. coli and P. aeruginosa that can use nitrate, nitrite, nitrogen oxide, and fumarate as alternative electron acceptors of the electron transport chain in anaerobic condition, susceptibility to aminoglycosides is restored to some extent (Campbell and Kadner, 1980; Bryan et al., 1979). 8 Figure 1.3 Aminoglycoside uptake and mode of action. Figure from (Krahn, 2012) 1) P. aeruginosa intact cytoplasmic and outer membrane in the absence of aminoglycosides 2) Ionic binding phase: Aminoglycosides bind to LPS molecules (red ovals). Aminoglycosides then displace Mg2+ and Ca2+ ions, which disrupts the cross-bridging of LPS molecules, damages the outer membrane and enhances its permeability. The outer membrane perturbation allows for the diffusion of the aminoglycosides in to the periplasm. 3) EDPI: Aminoglycoside traverse the CM using the energy provided by the membrane potential as well as the electron transport chain. Uptake of aminoglycosides is minimal at this point. 4) Ribosome-aminoglycoside interaction: Aminoglycosides bind to the ribosome and interfere with translation, leading to the production of mistranslated proteins (orange squiggles) 5) Mistranslated proteins insert the cytoplasmic membrane, perturbing its permeability. Small molecules, including aminoglycosides leak through the damaged membrane and enter the cytoplasm. This is the EDPII of aminoglycoside uptake. Reactive oxygen species (ROS) are produced as a result of cellular metabolic disruption, leading eventually to cell death. 9 10 EDPII is the accelerated form of EDPI stage (Nichols and Young, 1985) and it occurs after the interaction between aminoglycosides and the cytoplasmic aminoglycoside target, the ribosome (Bryan and Van Den Elzen, 1977). Once a small amount of aminoglycosides traverse the CM, the antibiotic binds to its target site on the ribosome, interferes with protein synthesis, causing the production of the aberrant proteins (to be described in details later in this section) (Figure 1.3 step 4). Some of these aberrant proteins that are produced as a result of aminoglycosides that have entered the cytoplasm via EDPI, insert the CM, perturbing it, increasing its permeability. This enhances further aminoglycoside uptake. An additional quantity of aminoglycosides are then transported through the damaged membrane, binding to their target sites, producing more aberrant proteins, and at last, leading to bacterial cell death (Davis et al., 1986; Taber et al., 1987) (Figure 1.3 step 5). The bacterial ribosome consists of two subunits with the relative sedimentation rate of 50S and 30S (Wilson et al., 2002). The 50S subunit, also known as the large subunit is comprised of two RNA molecules, the 23S and 5S rRNAs and 33 proteins. The 30S subunit, also known as the small subunit is comprised of a single RNA, 16S rRNA, and 20 to 21 proteins (Brodersen et al., 2002; Carter et al., 2000). The ribosome has three sites important for the translation process (Figure 1.4), the A-site, which is the site of entry for aminoacyl tRNA, the P-site, which is the site of the growing peptide chain, and the E-site where the newly synthesized polypeptide chain exits from (Green and and Noller, 1997). During protein synthesis, the information stored in the mRNA is decoded by the interaction on the A-site of the ribosome between a codon on the mRNA and an anti-codon on the tRNA (Ogle et al., 2003). The high fidelity of translation is achieved by the proper interaction between the cognate codons and anti-codons. Also, 16S rRNA located close to the A-site where the codon, anti-codon interaction occurs, ensures the accuracy 11 1) 2) 3) Figure 1.4 Ribosome sites of translation. Figure from (Freeman et al., 2008) 1) First aminoacyl tRNA carrying methionine resides on the P-site while the next aminoacyl tRNA resides on A-site, where its anti-codon pairs with the mRNA codon. 2) The amino acid on the tRNA residing on the P-site and the amino acid attached to the tRNA residing on the A-site are attached by a peptide bind and the amino acid on the tRNA residing on the P-site is separated from its tRNA. 3) Ribosome moves down the mRNA. The tRNA on the P-site now is transferred to the E-site to be exited. The tRNA with a peptide chain attached to is transferred to the P-site. The A-site is now empty and ready for the next aminoacyl tRNA to reside on. 12 of the translation by maintaining the specificity of this interaction. In the presence of a cognate aminoacyl tRNA, a conserved guanine residue G530 of 16S rRNA and two conserved adenine residues on helix H44 of 16S rRNA, A1492 and A1493 interact with each other. A1492 and A1493 usually face inwards on helix 44 of the 16S rRNA, but their interaction with G530 results in “flipped out” conformation of A1492 and A1493 residues of 16S rRNA which stabilizes the first two position base pairs of the cognate codon-anticodon interaction, which in turn promotes the incorporation of the correct amino acid in to the growing peptide chain (Ogle et al., 2003; Taliaferro and Farabaugh, 2007; Ogle et al., 2002). Noncognate aminoacyl tRNA, doesn’t allow for the interaction between G530 and A1492-A1493; therefore, no change in 16S rRNA A1492 and A1493 conformation occurs and the noncognate codon-anticodon interaction is destabilized. In the presence of aminoglycosides however, noncognate aminoacyl tRNA induces the interaction between G530 and 16S rRNA residues, which results in the “flipped-out” conformation of 16S rRNA A1492 and A1493, which in turn promotes the incorporation of the incorrect amino acid in to the growing peptide chain (Ogle et al., 2002; Taliaferro and Farabaugh, 2007). Most aminoglycosides, such as neomycin and gentamycin bind close to A1492 and A1493 regions of 16S rRNA (Moazed and Noller, 1987). Streptomycin on the other hand, binds to a site adjacent to the 2- deoxystreptamine aminoglycosides target site (Carter et al., 2000), specifically the phosphate backbone of 16S rRNA. The interaction between streptomycin and the ribosome results in stabilization of a high affinity for aminoacyl tRNA on the A-site (also called ram state). It also interferes with the proofreading state of translation (Leclerc et al., 1991; Thompson et al., 1981). Spectinomycin binds to helix34 of 16S rRNA, inhibiting the elongation factor G to catalyze the translocation of the tRNA and mRNA at the end of each round of polypeptide elongation, blocking the movement of the 30S ribosomal subunit, 13 and preventing A-site to P-site peptidyl-tRNA translocation (Bilgin et al., 1990). The exact mechanism by which aminoglycosides achieve their bactericidal effect has yet to be determined. As mentioned before, aminoglycosides interfere with protein synthesis and result in mistranslation and production of aberrant proteins (Jana and Deb, 2006; Shakil et al., 2008). Davis et al. in 1986 demonstrated that the aberrant proteins produced once the bacterial cell is exposed to aminoglycosides insert the CM, perturbing its integrity (Davis et al., 1986) and increasing its permeability. Uptake of aminoglycosides is then facilitated by the membrane damage, resulting in accumulation of aminoglycosides in the cytoplasm. One possible explanation for the bactericidal effects of aminoglycosides that depends on membrane perturbing effects of this antibiotic was stated by Kohanski et al. in 2007. Kohanski et al. (2007) stated that the three major classes of bactericidal antibiotics stimulate the production of highly deleterious hydroxyl radicals in Gram-negative and Gram-positive bacteria, which ultimately contribute to cell death (Kohanski et al., 2007). According to the model proposed by Kohanski et al. (2007), once the aberrant proteins insert the CM, they damage it and cause more aminoglycosides uptake. The membrane damage then activates an envelope stress response that has a variety of effects on metabolism, including hyper activity of the electron transport chain, resulting in excess oxidation of NADH, which consequently results in the excess generation of ROS, such as superoxide. Superoxide reacts with iron-sulfur clusters present in cellular proteins and generate toxic hydroxyl radicals via the Fenton reaction (Imlay et al., 1988). Hydroxyl radicals are proposed to contribute to cell death by damaging DNA, lipids, and proteins (Kohanski et al., 2007). Evidence was presented by Kohanski et al. (2007) to prove the accuracy of the model. Using iron chelators to block the Fenton reaction and hydroxyl radical production, Kohanski et al. (2007) showed that the bactericidal effect of the antibiotics (from all three major antibiotic 14 classes) was suppressed. Using thiourea, a hydroxyl radical scavenger, Kohanski et al. (2007) also showed suppression of antibiotic-mediated bacterial killing, consistent with the proposed model of aminoglycoside bactericidal effects. In 2010, Wang et al. also showed that mutations inactivating antioxidant defense mechanisms in bacteria increase bacteria susceptibility to antibiotics due to an increase in oxidative stress (Wang et al., 2010), again consistent with the proposed model of aminoglycoside bactericidal effects. Still, other studies by other groups questioned the involvement of ROS in antibiotic killing. Keren et al. in 2013 examined the effect of ROS on bacterial cell death using thiourea and stated that thiourea protected cells from antibiotics (from all three major antibiotic classes) at low concentration, but this effect was observed under both aerobic and anaerobic conditions, suggesting ROS do not play a role in bacterial killing by antibiotics (Keren et al., 2013). Liu and Imaly (2013) in a recent study showed that ampicillin, norfloxacin and kanamycin can kill bacteria in the absence of oxygen. They also showed that in E. coli mutants lacking catalases and peroxidases, which protect the cell from oxidative damage caused by ROS by decomposition of hydrogen peroxide to water and oxygen (Halliwell, 2005), only modestly makes the bacteria more susceptible to norfloxacin and has no effect on ampicillin or kanamycin bacterial susceptibility. None of these antibiotics increased the level of bacterial respiration or hydroxyl radical production (Liu and Imlay, 2013). If ROS were important in the bactericidal effect of aminoglycosides, mutants in peroxidases would have had a compromised defense mechanism, which would result in an increase in oxidative stress and aminoglycoside susceptibility; the result that was in contrary with what Liu and Imaly observed (2013). Recently in May 2014, Dwyer et al. confirmed their previous findings on the involvement of ROS in bactericidal effects of antibiotics. Using gentamicin, ampicillin and norfloxacin as representatives of the three major classes of antibiotics, they 15 demonstrated that the bactericidal antibiotics elevate oxygen consumption, leading to the production of more ROS, which increases oxidative stress which in turn damages DNA, lipids, and proteins in greater extent, leading to cell death (Kohanski et al., 2007). They further showed that overexpression of catalases or DNA mismatch repair enzymes, and anti-oxidant pretreatment, limit antibiotic lethality, by reducing the severe effects of ROS on bacterial cell, indicating that ROS causatively contribute to antibiotic killing. The killing efficacy of antibiotics was diminished under strict anaerobic conditions, but could be enhanced by exposure to molecular oxygen, suggesting that the presence of oxygen in the environment impacts the antibiotic lethality. Dwyer et al. recent work (2014) significantly confirms their previous findings, supporting an evolving, expanded model of antibiotic lethality. 1.2.3 Aminoglycosides resistance mechanisms in P. aeruginosa Resistance of P. aeruginosa to aminoglycosides was first reported in the 1960’s by Griffith et al. (1960 and 1966). Since then, there has been an ongoing resistance development against aminoglycosides in P. aeruginosa strains, especially those isolated from patients suffering from CF (Price et al., 1981; Saavedra et al., 1986). There are several resistance mechanisms by which P. aeruginosa fights against the bactericidal effects of aminoglycosides, including drug inactivation using modifying enzymes, drug target modification, impermeability resistance, biofilm formation, and mutational resistance, explained in details in the following sections. 1.2.3.1 Aminoglycoside modifying enzymes Common determinants of aminoglycoside resistance mechanism in P. aeruginosa are aminoglycoside modifying enzymes (AMEs) (Kettner et al., 1995; Miller et al., 1997; Miller et al., 1995). They chemically alter aminoglycosides and interfere with their binding to the ribosome (Llano-Sotelo et al., 2002), thereby blocking their activity. There are three families of 16 AMEs, aminoglycoside acetyltransferases (AACs), aminoglycoside phosphotransferases (APHs), and aminoglycoside nucleotidyltransferases (ANTs) (Wright, 1999). There are a variety of AMEs within each family, each differentiated on basis of the position on the aminoglycoside they modify. Thus, AAC (6’) is an acetyltransferase that modifies aminoglycosides at position 6’ (Figure 1.5). Different AMEs that modify an aminoglycoside at the same position are also known and differentiated within each family. AAC (6’)-I and AAC (6’)-II are differentiated based on their substrate profile, meaning they provide resistance to different antibiotics. While AAC (6’)-I confers resistance to amikacin, AAC (6’)-II confers resistance to kanamycin (Vakulenko and Mobashery, 2003). Individual enzymes that modify antibiotics at the same position and provide resistance to the same group of antibiotics but are encoded by different genes (Shaw et al., 1993a; Wright, 1999), are indicated by lower case letters. AAC (6’)-Ia, AAC (6’)-Ib, and AAC (6’)-Ic modify aminoglycosides at position 6’, providing resistance to the same antibiotics, including tobramycin, amikacin, netilmicin, kanamycin, and dibekacin, but are encoded by different genes. Figure 1.5 shows the modification site of all the P. aeruginosa modifying enzymes mentioned in this section. Table 1.1 summarized the information about all the P. aeruginosa modifying enzymes discussed in this section. AACs. Aminoglycoside acetyltransferases are comprised of four families AAC (1), AAC (3), AAC (6’), and AAC (2’), although AAC (6’) and AAC (3) are the most common AMEs used in P. aeruginosa clinical isolates (Galimand et al., 1993; Ramirez and Tolmasky, 2010; Poole, 2005a). They all substitute an amino group with an acetyl group given from acetyl-CoA, at positions 1 and 3 of 2-deoxystreptamine ring and positions 6’ and 2’ of 6- aminohexose ring respectively (Vakulenko and Mobashery, 2003). Aminoglycoside 6’ acetyltransferases [AAC (6’)] are the most frequently found acetyltransferases in P. aeruginosa. 17 ANT (3’) Figure 1.5 P. aeruginosa aminoglycoside modifying enzymes. Figure modified from (Kotra et al., 2000) The positions at which P. aeruginosa aminoglycoside modifying enzymes alter aminoglycosides (kanamycin is set as an example here) from all three classes, AAC, APH, and ANT are shown in the figure. The arrows show the site of modification by each specific enzyme. 18 Table 1.1 P. aeruginosa aminoglycoside modifying enzymes. Enzyme Resistance substance profile Reference AAC (6’) amikacin, tobramycin, kanamycin, Shaw et al., 1993a; Vanhoof et al., gentamicin 1998 gentamicin, paromomycin, Vliegenthart et al., 1991; Kettner tobramycin, and kanamycin et al., 1995; Rodríguez Esparragón AAC (3) et al., 2000; Miller et al., 1997 APH (3’) kanamycin, neomycin and Rodríguez Esparragón et al., 2000 streptomycin ANT (2”) Gentamicin, tobramycin MacLeod et al., 2000; Busch‐ Sorensen et al., 1996 ANT (4’) Kanamycin, tobramycin Sabtcheva et al., 2003; Jacoby et al., 1990; Shaw et al., 1993b ANT (3’) Streptomycin, spectinomycin 19 Shaw et al., 1991 (Vakulenko and Mobashery, 2003). AAC (6’) s modify and promote resistance to most of the clinically-important aminoglycosides. There are two classes of enzymes in this family of acetyltransferases, AAC (6’)-I, which confers resistance to amikacin, tobramycin, and kanamycin and AAC (6’)-II, which confers resistance to gentamicin and tobramycin (Vakulenko and Mobashery, 2003). Out of all AAC (6’) enzymes, AAC (6’)-Ib is the most common among Gram-negative microorganisms, and is a great contributor to aminoglycoside resistance in clinical infections caused by Gram–negative bacteria, especially P. aeruginosa (Shaw et al., 1993a; Vanhoof et al., 1998). Aminoglycoside 3 acetyltransferases, [AAC (3)] are also very common in providing resistance to aminoglycosides in P. aeruginosa. They are the second most common AAC enzymes used as resistance mechanisms in P. aeruginosa clinical isolates (Vakulenko and Mobashery, 2003). There are three classes of modifying enzymes in this family; AAC (3)-I, AAC (3)-II, and AAC (3)-III. Within AAC (3)-I class which promotes resistance to gentamicin, there are three sub classes of AAC (3)-Ia, AAC (3)-Ib, and AAC (3)-Ic. The genes for AAC (3)Ia and AAC (3)-Ib are found alone or together in up to 30% of the clinical isolates of Gramnegative bacteria (Miller et al., 1997; Shaw et al., 1992; Tenover et al., 1989; Wohlleben et al., 1989), typically on integrons in P. aeruginosa (Poirel et al., 2000) and are the major cause of gentamicin resistance. The other two classes of AAC (3) family, II (Kettner et al., 1995; Rodríguez Esparragón et al., 2000) and III (Vliegenthart et al., 1991), though less common, confer resistance to gentamicin, paromomycin, tobramycin, and kanamycin. APHs. Aminoglycoside phosphotransferases use a phosphoryl group from ATP to alter the chemical structure of aminoglycosides by attaching the phosphoryl group to specific hydroxyl groups. There are seven classes of APHs [APH (3’), APH (3”), APH (2”), APH (4), APH (7”), 20 APH (9), and APH (6)] (Shaw et al., 1993a), but only APH (3’) and APH (6) have been identified in P. aeruginosa. The most common APH enzyme present in P. aeruginosa is APH (3’) that confers resistance to kanamycin, neomycin and streptomycin by modifying the 3’-OH position of these antibiotics (Rodríguez Esparragón et al., 2000). APH (3’)-I and APH (3’)-II are both found commonly in clinical isolates of P. aeruginosam, typically on transposons (Oka et al., 1981; Beck et al., 1982), and provide resistance to kanamycin and neomycin (Rodríguez Esparragón et al., 2000; Shaw et al., 1991; Young et al., 1985). Perhaps P. aeruginosa high resistance to kanamycin and neomycin is due to the chromosomally encoded APH (3’)-IIb enzyme (Hachler et al., 1996). It is the most common APH (3’) enzyme, being chromosomally conserved, which contributes to intrinsic resistance. Other classes of APH (3’) provide resistance to aminoglycosides other than kanamycin and neomycin; APH (3’)-VI for example confers resistance to amikacin and isepamicin (Kettner et al., 1995; Lambert et al., 1994; Torres et al., 2000). ANTs. There are five classes of aminoglycoside adenyltransferases. They use ATP and modify aminoglycosides by substituting a hydroxyl group at positions 2”, 6’, 3, 4’, and 9 with an adenosine monophosphate (AMP) molecule (Houghton et al., 2010; Vakulenko and Mobashery, 2003). The most prevalent nucleotidyltransferase found in P. aeruginosa clinical isolates is the ANT (2”)-I enzyme that confers resistance to gentamicin (Busch‐Sorensen et al., 1996; Dornbusch and Hallander, 1980; Phillips et al., 1986; Reyonolds et al., 1976) as well as tobramycin (MacLeod et al., 2000). In addition to ANT (2”) class of adenyltransferases, ANT (4’) class also confers resistance to kanamycin and tobramycin in P. aeruginosa. Genes encoding ANT (4’)-IIa (Jacoby et al., 1990; Shaw et al., 1993b) and ANT (4’)-IIb (Sabtcheva et al., 2003) are located on both plasmids and the chromosome of P. aeruginosa isolates. Other adenyl21 transferases that confer resistance in P. aeruginosa include ANT (3’) that modifies streptomycin as well as spectinomycin (Shaw et al., 1991) and multiple variants of it is found encoded on integrons in P. aeruginosa (Ramirez and Tolmasky, 2010). 1.2.3.2 Target modification Although modifying enzymes are the most common resistance mechanism against aminoglycosides, target modification has also become an important aminoglycoside resistance mechanism in clinical isolates of P. aeruginosa since it confers resistance to all aminoglycosides by modifying their target site rather than conferring resistance to a particular aminoglycoside (Poole, 2005a). Target modification mechanism first appeared in organisms that naturally produced aminoglycosides, such as actinomycetes (Doi and Arakawa, 2007). Actinomycetes produce inactive acetylated or phosphorylated aminoglycosides which become active once the acetyl/ phosphoryl group is cleaved during or after their export from the cell by specific actinomycete-produced enzymes (Walsh, 2003; Tercero et al., 1996; Lacalle et al., 1993). To further aid in their resistance to their aminoglycoside products, actinomycetes produce methyltransferases that modify the 16S rRNA site of the ribosome, the target site of aminoglycosides. Methylation of the target site compromises the tight binding between aminoglycosides and the ribosome, protecting the cell from the antibiotic (Cundliffe, 1989). More recently, a gene encoding a16S rRNA methyltransferase, rmtA, was discovered in several P. aeruginosa clinical isolates in Japan (Yokoyama et al., 2003). RmtA conferred high-level resistance to all clinically-used aminoglycosides. In 2005 another 16S rRNA methyltransferase, RmtD was found in clinical isolates of P. aeruginosa in Brazil (Doi et al., 2007). RmtD conferred resistance to all 4, 6-deoxystreptamine aminoglycosides. The considerable primary 22 sequence similarity observed between the Rmt proteins and the 16S rRNA methylases of actinomycetes, as well as the GC content of the gene (55% similar to actinomycetes), suggests a possible gene transfer from the producing organism to P. aeruginosa (Magnet and Blanchard, 2005). In 2010, Gurung et al. reported the presence of ArmA methyltrasferase in 14 out of 100 multidrug-resistant P. aeruginosa strains from a Korean hospital. This was the first case of armA in P. aeruginosa (Gurung et al., 2010). In a study carried by Zhou et al. (2010), P. aeruginosa clinical isolates showed high resistance to aminoglycosides due to the presence of two 16S rRNA methyltransferases, ArmA and RmtB (Zhou et al., 2010). Out of 35 P. aeruginosa clinical isolates that showed high-level resistance to the tested aminoglycosides, including kanamycin, tobramycin, gentamicin, and amikacin, 31 strains were either armA (26 strains) or rmtB (5 strains)-positive (Zhou et al., 2010). 1.2.3.3 Impermeability resistance Impermeability aminoglycoside resistance was initially defined as reduced uptake and accumulation of all aminoglycosides due to change in outer membrane permeability (Yoshimura and Nikaido, 1982). Today impermeability aminoglycoside resistance is generally attributed to the activity of multidrug efflux pumps. Five classes of efflux pumps that provide resistance to antimicrobials have been described in bacteria (Li and Nikaido, 2009). However, members of the resistance nodulation division (RND) family appear to be the most significant contributors to antimicrobial resistance in P. aeruginosa (Poole, 2004; Poole, 2007). A P. aeruginosa efflux pump responsible for aminoglycoside resistance belongs to RND family and is called MexXYOprM (Mine et al., 1999). Like all the other members of RND family, MexXY-OprM consists of three components that include an inner membrane drug-proton antiporter (MexY), an outer membrane channel-forming protein (OprM) and a periplasmic link protein (MexX) that joins the 23 other two components (Poole, 2005b; Mine et al., 1999; Aires et al., 1999). MexX and MexY are encoded by the mexXY operon (Aires et al., 1999; Mine et al., 1999), but the outer membrane factor, OprM, is the product of the third gene of an operon encoding RND- type pump, MexABOprM (Aires et al., 1999; Masuda et al., 2000). MexXY- OprM promotes resistance to antimicrobials, such as β-lactams and fluoroquinolones, but is induced to great extent by those whose target site is the ribosome (Jeannot et al., 2005). Ribosome perturbation is a key to inducibility. Studies have shown an induced expression of mexXY once the bacterial cell is exposed to ribosome targeting drugs, such as aminoglycosides, tetracycline, and erythromycin that inhibit protein synthesis by interacting with the ribosome, as well as any ribosomal mutation that leads to the disruption of protein synthesis (Lau et al., 2012; Caughlan et al., 2009). Several studies have previously shown the contribution of MexXY efflux pump to aminoglycoside resistance in P. aeruginosa clinical isolates. In 2003, Sobel et al. showed that deletion of mexXY genes from 9 of the 14 isolates resulted in enhanced susceptibility to multiple aminoglycosides (up to 32-fold in some cases), confirming the contribution of this efflux system to the aminoglycoside resistance of these clinical isolates (Sobel et al., 2003). In 2009, Islam et al. also showed that MexXY efflux pump plays a significant role in P. aeruginosa aminoglycoside resistance. Out of 20 isolates, 17 showed MexY mRNA overproduction, correlated with increased resistance to aminoglycosides tested (amikacin and tobramycin) (Islam et al., 2009). The expression of mexXY is controlled by the MexZ repressor (Westbrock-Wadman et al., 1999), encoded by a gene located upstream of the mexXY operon (Aires et al., 1999; WestbrockWadman et al., 1999). Although inactivating mexZ mutations result in hyperexpression of MexXY- OprM (Islam et al., 2004; Smith et al., 2006) and these mutations are observed quite often in P. aeruginosa pan-aminoglycoside resistant (resistant to all aminoglycosides) clinical 24 isolates (Matsuo et al., 2004), there are studies that show MexXY-expressing aminoglycoside resistant clinical isolates lacked mutations in mexZ, suggesting aminoglycoside resistance in P. aeruginosa clinical isolates may be due to additional components (Sobel et al., 2003; WestbrockWadman et al., 1999). In 2006, Morita et al. showed that mutant strains disrupted in a gene called PA5471 were compromised for drug- inducible mexXY expression as well as MexXYmediated resistance (Morita et al., 2006). Morita et al. also showed that PA5471 was inducible by the same ribosome-targeting agents that induce mexXY expression. Introducing the excess amount of PA5471 to P. aeruginosa strain resulted in induced expression of mexXY and MexXY-mediated resistance in the absence of antibiotic exposure (Morita et al., 2006). The result of this experiment was consistent with the PA5471 directly or indirectly activating mexXY expression following its own up- regulation in response to antibiotic exposure. However, in mutant strains lacking mexZ, the requirement for PA5471 for mexXY expression and antimicrobial resistance was abolished, suggesting that PA5471 directly or indirectly modulates MexZ activity in effecting mexXY expression (Morita et al., 2006). In 2009, Yamamoto et al. further confirmed Morita et al. findings. Yamamoto et al. showed that when MexZ was expressed by a mexZ expression plasmid, the plasmid-borne MexZ repressed drug-induced MexX production, further confirming MexZ role as a mexXY operon repressor (Yamamoto et al., 2009). Yamamoto et al. also investigated the role of PA5471 protein in mexXY expression. PA5471 was expressed by a PA5471 expression plasmid. The plasmid-borne PA5471 induced MexX as well as MexZ production, but further experiments revealed that the interaction between PA5471 and MexZ reduced MexZ DNA binding ability, leading to mexXY expression (Yamamoto et al., 2009). In 2013, Hay et al. showed that in a strain lacking PA5471 (MexZ+, PA5471-), induction of mexXY by a model ribosome-disrupting compound, spectinomycin, the 25 agent observed to most strongly induce this efflux operon (C.H.F. Lau, unpublished), was wholly compromised. However, exposure of the ΔmexZ mutant strain (MexZ-, PA5471+) to spectinomycin enhanced mexXY expression to levels seen for spectinomycin-exposed wild-type strain (MexZ+, PA5471+). Elimination of PA5471 in ΔmexZ mutant strain (MexZ-, PA5471-) had no effect on spectinomycin-inducible mexXY expression. These experiments showed that PA5471 was required for antimicrobial-inducible mexXY expression only when MexZ repressor was present, indicating that PA5471 acts as MexZ antirepressor. Therefore, PA5471 was further named armZ, (antirepressor MexZ) (Hay et al., 2013). 1.2.3.4 Biofilm P. aeruginosa is an opportunistic human pathogen that causes a variety of diseases, including chronic pulmonary infection in CF patients, characterized by the formation of biofilms (Wagner and Iglewski, 2008; Davies and Bilton, 2009). Biofilms are surface-attached structures in which bacteria are embedded in a matrix comprised of polysaccharide, protein, and DNA (Lopez et al., 2010). The resistance characteristics of the cells within the biofilm depend on the position of the cells in the matrix (Werner et al., 2004). Those that are attached to the surface have better access to nutrients while those embedded deeply in the matrix have less access to nutrients and oxygen; therefore, those cells located in deeper layers of the biofilm are known to be less metabolically active (Werner et al., 2004; Pamp et al., 2008). It is generally believed that cells on the biofilm periphery are more susceptible to antimicrobials, including aminoglycosides since they have adequate supplies of nutrients and are metabolically active while cells located in deeper layers are more resistant to antimicrobial agents since they most likely metabolize more slowly and have limited access to nutrients and oxygen. As many antimicrobial agents require actively metabolizing cells to be effective (Pamp et al., 2008), the presence of slow growing or dormant 26 cells is thought to represent a resistant population. Also, since cells residing in deeper layers of the biofilm have less access to oxygen, they mimic an anaerobic condition and therefore, they are more resistant to aminoglycosides once compared to the cells located on the surface of the biofilms (Hassett et al., 2009). The position of the cells within the matrix, and their accessibility to oxygen and nutrients are some of the reasons the biofilm is thought to be more resistant to antibiotics, including aminoglycosides. Another possible reason for increased antibiotic resistance in biofilm is poor diffusion of antibiotics through the biofilm polysaccharide matrix. Presence of exopolysaccharide alginate in large quantities in P. aeruginosa biofilms, have been shown to inhibit easy diffusion of aminoglycosides to the bacterial cell, leading to reduced access of aminoglycosides to their target site, and an increase in resistance (Hatch and Schiller, 1998; Nichols et al., 1988). The presence of extracellular DNA in the matrix of the biofilm may also contribute to an increase in resistance in biofilms. Negatively-charged extracellular DNA may compete with the negatively-charged portion of the bacterial membrane in binding to the cationic aminoglycosides, thus increasing aminoglycoside resistance in biofilms due to reduced access to the bacteria within the biofilm (Mulcahy et al., 2008). Another explanation for increased aminoglycoside resistance in biofilms is the presence of highly resistant subpopulation of dormant cells called persisters (Lewis, 2010; Lewis, 2008). Persisters are dormant, not growing cells present in both biofilm and planktonic cell population. They are the only cells to survive treatment with high doses of bactericidal antibiotics, mainly due to the antibiotic target sites being inactive in persister stage. Persister cells show a phenotypic variation that is more resistant to antimicrobial agents than the rest of the biofilm, but they are genetically identical to the rest of the biofilm population (Mulcahy et al., 2010). Aminoglycosides induce biofilm formation in P. aeruginosa by a gene called arr, encoding 27 an aminoglycoside response regulator, whose lack compromises biofilm resistance to aminoglycosides (Hoffman et al., 2005). arr encodes phosphodiesterase that impacts the levels of a second messenger (c-di-GMP) known to influence biofilm formation. Another gene needed in some strains of P. aeruginosa for biofilm-specific resistance to aminoglycosides is ndvB (Mah et al., 2003). ndvB is involved in the synthesis of periplasmic glucans. By binding to aminoglycosides, glucans prevent this antibiotic to reach its target, making the biofilm more resistant to aminoglycosides. 1.2.3.5 Mutational resistance P. aeruginosa is a metabolically versatile bacterium capable of living in multiple ecological niches (Silby et al., 2011), including the airways of CF patients (Smith et al., 2006). One of the challenges P. aeruginosa faces in the CF lung environment is intensive antibiotic therapy. Causing chronic pulmonary infection in the lungs of CF patients (Lyczak et al., 2002; Govan and Deretic, 1996; Gibson et al., 2003), P. aeruginosa is constantly exposed to antibiotics, most commonly aminoglycosides. Over time, P. aeruginosa adapts to the environment and exhibits acquired resistance against the specific aminoglycoside antibiotics it has been exposed to (Smith et al., 2006; Macia et al., 2005). Some environments, particularly lungs are rich in ROS (Lagrange‐Puget et al., 2004; Wood et al., 2001) and it is well known today that exposure to ROS have deleterious effects on molecules, such as DNA, lipids, and proteins (Dwyer et al., 2009; Kohanski et al., 2010). ROS are also generally known to be mutagenic (i.e. cause mutation), an example of such being mutations in DNA repair genes, which is one of the most common mutations occurring in some strains of P. aeruginosa (Oliver and Mena, 2010). DNA repair genes mutations can further develop multiple secondary mutations (i.e. hypermutation) (Breidenstein et al., 2011), which can occur in any of the P. aeruginosa genes, leading to an 28 increase in mutation frequency. In the presence of antibiotics, including aminoglycosides, drugresistant mutations may be selected for, leading to an increase in antibiotic resistance in the hypermutator strains (Oliver et al., 2002; Schurek et al., 2008) . In addition to hypermutation, single mutation in genes that enable many of the key physiological events in activity or uptake of aminoglycosides contribute to resistance to this class of antibiotic. Transposon mutagenesis studies, carried in different P. aeruginosa strains have identified genes whose disruption contribute significantly to aminoglycoside resistance (Dotsch et al., 2009; Fajardo et al., 2008; Schurek et al., 2008; El'Garch et al., 2007; Krahn et al., 2012). Investigated by El’Garch et al. (2007), screen of a random transposon insertion mutants in P. aeruginosa identified four genes (galU, nuoG, mexZ, and rplY) whose disruption resulted in increased resistance to aminoglycosides. GalU is important for the complete formation of LPS molecule and since LPS is involved in ionic binding phase of aminoglycoside uptake (Taber et al., 1987), it is reasonable that galU disruption contributes to aminoglycoside resistance. NuoG contributes to proton motive force across the CM. EDPI and EDPII steps of aminoglycoside uptake require energy provided by proton motive force; therefore, it would be reasonable if nuoG disruption leads to alteration in aminoglycoside uptake and its activity (Bryan and Van Den Elzen, 1977; Campbell and Kadner, 1980). Mutation in mexZ, encoding a transcriptional repressor of mexXY, also contributes to aminoglycoside resistance as previously demonstrated (Matsuo et al., 2004; Westbrock-Wadman et al., 1999). Mutation in rplY, encoding a ribosomal protein, would also be associated with aminoglycoside resistance as ribosome is the aminoglycosides’ target site (Vakulenko and Mobashery, 2003) and alterations in the ribosome structure can also affect aminoglycoside efficacy. In another study carried by Schurek et al. (2008), random transposon insertion in P. aeruginosa has indicated mutations in genes related to 29 NADH reduction, including the nuo and nqr operons, as well as PA3493, encoding the putative NADH: ubiquinone oxidoreductase (Schurek et al., 2008). Several aminoglycoside-resistant cytochrome mutants (sucC, tatC, and ccmF) have also been reported (Schurek et al., 2008). NADH and cytochrome mutations are important since they are both involved in electron transport chain that provides energy needed for EDPI and EDPII steps of aminoglycoside uptake (Taber et al., 1987; Bryan and Kwan, 1983). 1.3 Statement of purpose P. aeruginosa gradually develops resistance through the acquisition of mutations in genes that are intrinsically involved in resistance when it is constantly exposed to a certain type of antibiotic, such as constant exposure to aminoglycosides in lungs of CF patients. In 2012, Krahn et al. performed a random mutagenesis study using a P. aeruginosa pan-aminoglycoside clinical isolate to identify genes that are intrinsically involved in aminoglycoside resistance (some of them are listed in Table1.2). A transposon insertion in either of two genes, PA2797 and PA2798, identified as a two component system, resulted in significant (8-16 fold) decrease in aminoglycoside resistance. Further microarray analysis of a P. aeruginosa K767 ΔPA2798 mutant showed a change in the expression of several genes whose products’ possible involvement in PA2797-PA2798- depleted aminoglycoside resistance was the subject of this study (some of them are listed in Table1.3). Since the expression of genes involved in the general stress response (rpoS), anaerobic respiration (anr), oxidative stress response (lexA and recN), as well as heat shock-related genes and a ribosomal protein operon (rpsP operon) was altered more than other genes in the mutant strain, pathways that this genes were involved in were selected for further investigation. The purpose of this study was to define the mechanism by which PA2797PA2798 promotes aminoglycoside resistance in P. aeruginosa such that its loss compromises 30 aminoglycoside resistance. A better understanding of the exact role of PA2797-PA2798 in aminoglycoside resistance may provide insight in to aminoglycoside resistance in P. aeruginosa, suggesting ways of avoiding it. 31 Table 1.2 Genes whose disruption by a transposon insertion decreased aminoglycoside resistance Gene Known/predicted function of the product lptA Lipid biosynthesis faoA Lipid metabolism pstB Phosphate uptake amgRS Two component regulatory system PA2797-PA2798 Two component regulatory system PA0392 Unknown function 32 Table 1.3 Genes whose expression was changed in K767 ΔPA2798 and were investigated in this study Gene Known/predicted function of product norB Nitric-oxide reductase norC Nitrogen metabolism dnaJ Heat shock protein grpE Heat shock protein htpG Heat shock protein lexA SOS response repressor recN DNA repair protein rpsP Ribosomal protein rimM Ribosomal protein trmD Ribosomal protein rplS Ribosomal protein 33 Materials and Methods 2.1 Bacterial strains and growth conditions The bacterial strains and plasmids used in this study are listed in Tables 2.1 and 2.2 respectively. Cultures of P. aeruginosa as well as E. coli were grown in Miller’s Luria broth, [Difco], with 2.5 g NaCl added per liter of growth medium (collectively referred to as L-broth), at 37°C shaking continuously, and were plated on L-broth with 1.5% (wt/vol) agar [Difco] (collectively referred to as L-agar); antibiotics were added as needed to the growth medium for the purpose of maintaining plasmids. Tetracycline (TET) was used to maintain plasmids pEX18Tc, and pRK415, and their derivatives in E. coli strains (10 µg/ml) and P. aeruginosa strains (50 µg/ml). Ampicillin and carbenicillin were used to maintain the plasmid pUCP19 and its derivatives in E. coli strains (100 µg/ml) and P. aeruginosa strains (400 µg/ml). All bacterial strains were grown in L-broth, incubated at 37°C for 18hrs with shaking (90 rpm). 2.2 DNA protocols Standard protocols were used for colony polymerase chain reaction (PCR), restriction endonuclease digestions, ligations, transformations, and agarose gel electrophoresis as stated in Sambrook and Russell (2001). CaCl2-competent E. coli (Sambrook et al., 2001) and electrocompetent P. aeruginosa (Choi et al., 2006; Masuda and Ohya, 1992) cells were prepared as previously described. Plasmids were maintained in E. coli strain DH5α and isolated using the GeneJETTM Plasmid Miniprep Kit according to the protocol suggested by the manufacturer (Fermentas, Inc., Burlington, ON). Plasmid pRK415 and its derivatives were maintained in E. coli strain DH5α and isolated using the QIAfilterTM Plasmid Midi Kit according to the 34 Table2.1 Bacterial strains used in this study Strain Description Source or Reference K3162 K3163 K3627 K3628 PAO1 wild-type strain K767Δanr K767Δ rpoS K767ΔPA2798 K767ΔPA2797 K767ΔPA2798ΔrpoS K767ΔPA2798Δanr Masuda and Ohya, 1992 This study Krahn et al., 2012 Krahn et al., 2012 Krahn et al., 2012 This study This study PAO1 PAO-MW20 Wild-type strain PAO1 rpoS-Gm Whiteley et al., 2000 Whiteley et al., 2000 Φ80 ΔlacZΔM15 endA1recA1hsdR17 (rK-mL+) supE44 thi-1 gyrA96 relA1 F-Δ(lacZYA-argF)U169 Ausubel et al., 2002 Donor strain used to promote transfer of pEX18Tc derivatives into P. aeruginosa; thi pro hsdR recA Tra+ Simon et al., 1983 P. aeruginosa K767 K3616 E. coli DH5α S17-1 35 Table 2.2 Plasmids used in this study Plasmid Description Source or Reference pEX18Tc Broad-host-range gene replacement r vector; sacB, Tc Hoang et al., 199 pRK415 Broad-host-range cloning vector; Plac-MCS Tcr Keen et al., 1988 pUCP19 Cloning vector; Apr Schweizer, 1991 pRA001 pEX18Tc::Δanr This study pRA002 pUCP19::PA2797 This study pRA003 pRK415::anr This study pRA004 pUCP19::rpoS This study pRA005 pUCP19::anr This study pRA006 pRK415::rpoS This study pRA007 pRK415::PA2798 This study pRA008 pUCP19::PA2798 This study pRA009 pRK415::rpsP operon This study pRA010 pUCP19::rpsP operon This study pCG008 pEX18Tc::ΔPA2797 Krahn et al., 2012 pCG009 pEX18Tc::ΔPA2798 Krahn et al., 2012 pTLK001 pEX18Tc::ΔrpoS T. L. Krahn 36 protocol provided by the manufacturer (Qiagen, Mississauga, ON). Chromosomal DNA was extracted from P. aeruginosa using a modified protocol of DNeasy® Blood & Tissue Kit (Qiagen, Mississauga, ON). One milliliter of the overnight culture was harvested in a 1.5 ml micro-centrifuge tube by centrifuging at 13000 rpm for 2 minutes. The supernatant was removed and discarded, and the pellet was re-suspended in 180 µl of buffer ATL. Proteinase K (20 µl of 20 mg/ml) was added and mixed by vortexing. The mixture was then incubated at 55°C for 20 minutes. RNase A (40 µl of 10 mg/ml) was then added and the mixture was incubated at room temperature for 2 minutes. Two hundred microliter of buffer AL was then added to the mixture and incubated for 10 minutes at 70°C. Two hundred microliter of 95% (vol/vol) ethanol was then added to the mixture and was mixed thoroughly by vortexing. The mixture was added to the DNeasy Mini spin column and centrifuged at 8000 rpm for 1 minute. The flow-through was then discarded. Five hundred microliter buffer AW1 was added to the spin column and the mixture was then centrifuged at 8000 rpm for 1 minute. The flow-through was then discarded. Five hundred microliter of buffer AW2 was added to the spin column and the mixture was centrifuged at 13000 rpm for 3 minutes. The flow-through was again discarded. The column was placed in a fresh 1.5 ml micro-centrifuge tube and 200 µl of buffer AE was added directly on to the DNeasy membrane. The mixture was incubated at room temperature for 1 minute and then centrifuged for 1 minute at 8000 rpm to elute the chromosomal DNA which was then stored at 4°C for further use in the experiments. PCR products and restriction endonuclease digestion products requiring purification were purified using the Promega Wizard SV Gel and PCR Clean Up System (Promega Corp., Madison, WI) according to a protocol provided by the manufacturer. DNA sequencing was performed by ACGT Corporation (Toronto, ON) and oligonucleotides were synthesized by Integrated DNA Technologies (IDT, Coralville, IA). Primers used in this 37 study are all listed in Tables 2.3 to 2.5. 2.3 Construction of genetic deletion in P. aeruginosa Derivatives of P. aeruginosa strains with deletion of genes of interest were generated as per methodology described previously (Sobel et al., 2003). Gene deletions were constructed by amplifying via PCR, 1-kb fragment upstream and downstream of the sequences being deleted. To introduce gene deletion in P. aeruginosa, deletions were first constructed in plasmid pEX18Tc (Table 2.2) and introduced in to E. coli DH5α. Plasmids pEX18Tc harboring the deletion construct were then transformed in to E. coli S-17 and delivered to the P. aeruginosa chromosome via homologous recombination. E. coli S-17 containing the plasmid with the relevant genetic construct, also known as the donor strain, was grown overnight in L-broth supplemented with TET 5 µg/ml for plasmid maintenance, at 37°C, shaking at 2000 rpm. The parent P. aeruginosa K767 strain, the recipient, was grown overnight in L-broth at 42°C, static. Conjugation and control plates (donor and recipient) were prepared and grown overnight at 37°C. Colonies from the conjugation plates as well as the control plates were diluted and plated using glass beads on L-agar plates containing TET 50 µg/ml (except P. aeruginosa K3627 and P. aeruginosa K3628 where TET 75 µg/ml was used) and chloramphenicol (CAM) 5 µg/ml (to counter select E. coli S-17). Colonies grown on the conjugation plates were then streaked on Lagar supplemented with 10% (wt/vol) sucrose, and grown overnight at 37°C to select against cells retaining the plasmid. Sucrose-resistant colonies were screened for the appropriate deletion using colony PCR. Colony PCR was carried out using the respective upstream forward and downstream reverse primers for each deletion (Table 2.3). Each 10 μl PCR reaction contained 30 pmol of each of the forward and reverse primers, 10% DMSO (vol/vol), 1 μl of thermopol (TP) buffer, 0.5 U Taq polymerase, and 0.2 mM of each deoxynucleoside triphosphate (dNTP). 38 Table 2.3 Primers used in gene deletion (5’ to 3’ order) Primer Sequence Source anr Up For CTAGGGTACCGACTACCAGACCAGCC This study anr Up Rev CTAGGGATCCCATTGAGGGGTCCTTG This study anr Dn For CTAGGGATCCGAAGTGCACATCCTCG This study anr Dn Rev CTAGAAGCTTGGAAATTGGCGCGAAC This study PA2798 Up-F GACTGAATTCCCGTACGTGATGCTGCCGTT Krahn et al., 2012 PA2798 Down-R GACTAAGCTTGGTCTCGCGCATCTATCGCT Krahn et al., 2012 39 Table 02.4 Primers used in gene cloning (5’ to 3’ order) Primer Sequence Source anr For ACGCAAGCTTTTACCCTTACTCCTGTT This study anr Rev ACGAGGTACCTCATGGAAGATCCTGG This study rpoS For ACATAAGCTTCACATCATGTAGGTGAG This study rpoS Rev ACATGGTACCCCGGGTCTTCAGTGGG This study rpsP operon For ACGCAAGCTTGAATATGCGGCCTTTCG This study rpsP operon Rev ACGCGAATTCGAAAGGCGAACGCCAG This study 40 Table 02.5 Primers used in q-RT PCR (5’ to 3’ order) Primer Sequence Source recN For AGAAGACCCTGAGCAACG This study recN Rev CATTGAGCGCGGAAAGGA This study lexA For TCTGGCTGCTGGCGGAAAC This study lexA Rev GACGCTCAAGCCTTCGATGA This study pslA For CGCTTCCGCATCATGTTC This study pslA Rev GCTGAGGTAGGGAAACAG This study rpsP For GTGACCAACAGCCGCAATG This study rpsP Rev CTTGAGCAGCTGAGCAAC This study norB For GAAGGCGTGTGGGAACTGA This study norB Rev GCCACTTCTCGATCACCTCG This study qPCR-rpoD-F ATCCTGCGCAACCAGCAGAA Lau et al., 2012 qPCr- rpoD-R TCGACATCGCGCGGTTGATT Lau et al., 2012 41 A standard colony PCR protocol was used for each deletion unless otherwise stated. Samples were heated for 3 minutes at 95°C, followed by 34 cycles of 45 seconds at 95°C, 30 seconds at 55°C (annealing temperature), 1 minute and 45 seconds (extension time) at 72°C, before finishing with 5 minutes at 75°C. 2.3.1 Deletion of anr in P. aeruginosa K767 For P. aeruginosa K767 Δanr, the upstream fragment was amplified using primers anr Up For, and Up Rev (Table 2.3) and the downstream fragment was amplified using primers, anr Dn For, and Dn Rev (Table 2.3). KpnI- BamH1 and BamH1-HindIII restriction sites were included in upstream and downstream primers respectively. The 50 μl PCR reaction mixtures contained 1 μg of chromosomal P. aeruginosa K767 DNA as template, 0.2 μM of each primer, 0.2 mM of each dNTP, 1x Pfu (+Mg) buffer, and 1.25 U Pfu DNA polymerase (Promega). PCR amplification was initiated with denaturation step at 98°C for 3 minutes. The mixture was subject to 30 cycles of heating at 98°C for 30 seconds, 64.5°C (annealing temperature) for 30 seconds, and 72°C for 1 minute (extension time), before finishing with 5 minutes incubation at 72°C. The constructed plasmid pRA001 (pEX18Tc:: Δanr) was used to construct an in-frame deletion of anr gene in P. aeruginosa K767 strain via homologous recombination as stated in section 2.3. Colony PCR was carried out using the primers anr Up For and anr Dn Rev (Table 2.3). The standard colony PCR condition mentioned in section 2.3 was used with an annealing temperature at 58.6°C and extension time of 3 minutes and 15 seconds. 2.3.2 Deletion of PA2798 in P. aeruginosa The previously constructed plasmid pCG009 (pEX18Tc:: ΔPA2798) was used to introduce PA2798 deletion in to strains K3616 carrying an in-frame deletion of anr and K3633 carrying an in-frame deletion of rpoS as per the protocol described in section 2.3. Colonies of potential Δanr 42 derivatives of K3162 as well as ΔrpoS derivatives of K3162 were screened via colony PCR. Colony PCR was carried out using the primers PA2798 Up-F and PA2798 Down-R (Table 2.3). The standard colony PCR condition mentioned in section 2.3 was used with an annealing temperature at 63.1°C for 45 seconds. 2.4 Assessment of antimicrobial susceptibility Antimicrobial susceptibility was assessed in wild-type and mutant strains of P. aeruginosa using a two-fold serial dilution technique (Amsterdam, 1996), using antibiotic stocks prepared at 50 mg/ml (paromomycin, spectinomycin, streptomycin, and kanamycin) and 5mg/ml (gentamicin, tobramycin, and amikacin). Fifty microliter of L-broth was first added to a 96-well microtiter plate. Antibiotics were then serially diluted two-fold in the L-broth across 96-well plates, 50 μl per well, followed by addition of 50 μl of P. aeruginosa strains diluted 1/2000 from overnight cultures. Plates were incubated at 37°C overnight for 20hrs and minimum concentration of antibiotics inhibiting visible growth (MIC) was recorded. 2.5 Growth assay Growth of P. aeruginosa strains was assessed at 30°C, 37°C, and 42°C using a growth assay. Briefly, Overnight cultures of P. aeruginosa were diluted into fresh L-broth (1/50). With initial optimal density at 600nm (OD600) of 0.05, strains K767, K3162 and K3163 were incubated at 30°C, 37°C, and 42°C shaking (200 rpm). Growth was assessed by measuring the optimal density of each strain at 1hr interval for 6hrs. 2.6 Gene cloning Various genes were amplified from P. aeruginosa K767 chromosomal DNA using primers tagged with restriction sites (Table2.4) to facilitate their cloning in to plasmids pRK415 (low copy 43 number plasmid) and pUCP19 (high copy number plasmid). Genes were amplified via PCR, including 50 bp fragments upstream and downstream of the sequences being cloned. The 50 μl PCR reaction mixtures contained 1 μg of chromosomal P. aeruginosa K767 DNA as template, 0.2 μM of each primer, 0.2 mM of each dNTP, 1x Pfu (+Mg) buffer, and 1.25 U Pfu DNA polymerase (Promega). PCR amplification was initiated with denaturation step at 98°C for 3 minutes. The mixture was subject to 30 cycles of heating at 98°C for 30 seconds, 64.5°C (annealing temperature) for 30 seconds, and 72°C for 1 minute (extension time), before finishing with 5 minutes incubation at 72°C. Plasmids carrying the gene of interest were first transformed in to CaCl2-competent E. coli DH5α (Sambrook et al., 2001). To ensure that no error had been introduced during PCR, cloned genes were sent for sequence check by ACGT Corporation (Toronto, ON). Plasmids carrying the gene of interest were then electroporated in to P. aeruginosa (Choi, 2005). 2.6.1 Cloning anr anr gene was amplified using the primers anr For and anr Rev (Table2.4). HindIII and KpnI restriction sites were included in forward and reverse primers’ sequence respectively. anr was amplified via PCR, as per protocol mentioned in section 2.6 with an annealing temperature of 68.1°C and extension time of 1 minute. 2.6.2 Cloning rpoS rpoS gene was amplified using the primers rpoS For and rpoS Rev (Table2.4). HindIII and KpnI restriction sites were included in forward and reverse primers’ sequence respectively. rpoS was amplified via PCR, as per protocol mentioned in section 2.6 with an annealing temperature of 60.0°C and extension time of 1 minute and 30 seconds. 44 2.6.3 Cloning rpsP operon rpsP operon consists of four genes: rpsP, rim, trmD, and rplS. Primers were designed to cover sequences of all four genes. rpsP operon was amplified using the primers rpsP operon For and rpsP operon Rev (Table2.4). HindIII and EcoRI restriction sites were included in forward and reverse primers’ sequence respectively. rpsP operon was amplified via PCR, as per protocol mentioned in section 2.6 with an annealing temperature of 69.9°C and extension time of 2 minutes and 15 seconds. 2.7 Quantitative real-time PCR Gene expression was assessed in this study using quantitative real-time PCR (q-RT-PCR) as previously described by Lau et al. (Lau et al., 2012). Briefly, overnight cultures were grown in Lbroth, supplemented with the antibiotics to maintain plasmids as needed at 37°C with shaking (200 rpm). Subcultures of 1/50 dilution were prepared and incubated at 37°C until the cell density reached 0.6-0.8 (OD600). Subcultures of 1.5 ml were then subject to RNA extraction using a High Pure RNA Isolation Kit (Roche Diagnostics Canada, Mississauga, ON) as per manufacturer’s instructions. Remaining traces of DNA in the isolated RNA samples were eliminated by a 30 minute treatment with Turbo DNA-free DNase (Applied Biosystems Canada, Streetsville, ON) again as described by the manufacturer. RNA yields were typically 3 to 9 µg RNA per extraction. Extracted RNA was then converted to cDNA using the iScript cDNA Synthesis Kit according to the instructions provided by the manufacturer (Bio-Rad, Mississauga, ON). q-RT PCR was performed using the Bio-Rad CFX 96TM Real-Time PCR Detection System (Bio-Rad, Mississauga, ON). The abundance of target and reference gene mRNA was measured in a 20 µl reaction mixture containing 5 µl of a 200-fold diluted cDNA template (an amount corresponding to 1.25 ng of total RNA), a 0.3 mM (pslA) or 0.6 mM (recN, lexA, norB, rpsP, 45 rpoD) concentration of each primer (Table 2.5), and 10 µl of the SsoFast Eva Green supermix (Bio-Rad). The q-RT PCR conditions were as follows: an initial 3-minute denaturation at 95°C, 40 cycles of 10 seconds denaturation at 95°C, 15 seconds annealing at 60°C, finishing with 10 seconds at 95°C. To verify the specificity of each amplification reaction, the melting curve profile of the resultant amplicons was determined over a range of 65°C to 95°C. q-RT PCR condition for all genes assessed were as per protocol described above except norB with annealing time of 30 seconds. A housekeeping sigma factor gene, rpoD, was used in each experiment as a reference gene and the expression of each gene was normalized to rpoD expression as previously recommended by Savli et al.(Savli et al., 2003). 46 Results 3.1 The connection between RpoS and P. aeruginosa aminoglycoside resistance According to Krahn et al. (Krahn et al., 2012), loss of the PA2798 gene causes the cells to become less resistant to aminoglycosides (8-16 fold). With 46% similarity to RssB (31.67% identity; 5.67% conserved changes) (by protein data bank; http://www.rcsb.org), an anti-RpoS anti-sigma factor in E. coli, it was hypothesized that in P. aeruginosa, PA2798 might affect aminoglycoside resistance through RpoS. RpoS is a master regulator of the general stress response mechanism in E. coli and its activity is repressed by RssB (Hengge-Aronis, 2002; Klauck et al., 2001). In P. aeruginosa, however, the role of RpoS is less well defined; it acts as an alternative sigma factor and is involved in stress responses such as nutritional stress response, heat shock stress response, envelope stress response, and oxidative stress response (reviewed in Poole 2012) as P. aeruginosa rpoS mutants show increased susceptibility to carbon starvation, heat shock, increased osmolarity, and low pH respectively (Schuster et al., 2004; Jorgensen et al., 1999; Suh et al., 1999). Aminoglycosides are cell damaging antibiotics and activate a variety of stress response mechanisms, including heat shock stress response (Kindrachuk et al., 2011) envelope stress response (Lau et al., 2013) and oxidative stress response (Fraud and Poole, 2011) once they enter the bacterial cell; therefore, it might be possible that RpoS affects stress response mechanisms being initiated upon aminoglycoside entry and action on the cell. There were two possibilities as to how RpoS might be connected to aminoglycoside resistance: 1) PA2797-PA2798 positively affects RpoS activity and then RpoS positively affects aminoglycoside resistance (i.e. loss of PA2797-PA2798 compromises RpoS activity and so, reduces aminoglycoside resistance); 2) PA2797-PA2798 negatively affects RpoS activity with 47 RpoS negatively affecting aminoglycoside resistance (i.e. loss of PA2797-PA2798 increases RpoS activity which reduces aminoglycoside resistance). To assess the first possibility, an attempt was made to delete the rpoS gene in wild-type P. aeruginosa strain K767. If the first hypothesis was true, then knocking out rpoS in the wild-type strain would mimic what occurred when PA2798 was knocked out in P. aeruginosa; P. aeruginosa lacking rpoS would yield a decrease in aminoglycoside resistance. Initially, an available P. aeruginosa strain with a mutation in the rpoS gene (Whiteley et al., 2000) was used to study the impact of rpoS loss on aminoglycoside resistance. As shown in Table 3.1, no change in resistance was observed in the rpoS mutant strain as compared with its wild-type strain. Although there was a modest 2-fold increase in paromomycin resistance in the rpoS mutant, this did not support the initial hypothesis. While a connection between RpoS and aminoglycoside resistance was not observed, it was noted that the wild-type parent strain of the rpoS mutant was different from the wild-type K767 strain in which the connection between PA2797-PA2798 and aminoglycoside resistance was previously observed (Krahn et al., 2012). Thus, a derivative of P. aeruginosa K767 with an in-frame deletion of the rpoS gene (generated by T. Krahn, unpublished) was assessed for change in aminoglycoside resistance. As shown in Table 3.1, no change in resistance was observed in P. aeruginosa K767 ΔrpoS, as compared with the wild-type parent strain. To confirm that P. aeruginosa K767 ΔrpoS was truly lacking RpoS function (i.e. whether the in-frame deletion was successful), the expression of pslA, an RpoS target gene, was assessed in both P. aeruginosa K767 and P. aeruginosa K767 ΔrpoS. As shown in Figure 3.1, there was a 2- fold decrease in pslA expression in P. aeruginosa K767 ΔrpoS, confirming the loss of RpoS function in this strain. Thus, loss of RpoS function does not decrease P. aeruginosa aminoglycoside resistance. Therefore, the first hypothesis PA2797-PA2798 positively affects RpoS function and RpoS 48 Table 3.1 Impact of loss of rpoS on aminoglycoside resistance in P. aeruginosa Strain rpoSa Minimum Inhibitory Concentration (µg/ml) b PAR SPC KAN STR AMI TOB PAO1 + 256 256 64 16 2 1 PAO-MW20 - 128 256 64 16 2 1 K767 + 128 512 64 16 2 0.5 K3633 - 128 512 64 16 2 0.5 a + ,wild-type; - , ΔrpoS (for K3633) and rpoS::Gm (where Gm= gentamicin cassette inserted in to rpoS) (for PAO-MW20) b abbreviations used: PAR, paromomycin; SPC, spectinomycin; KAN, kanamycin; STR, streptomycin; AMI, amikacin; TOB, tobramycin 49 pslA expression (fold-change) 1.2 1 0.8 0.6 0.4 0.2 0 RpoS+ RpoS- Figure 3.1 Impact of loss of rpoS on the expression of pslA. The expression of pslA was assessed in wild-type P. aeruginosa K767 strain (RpoS+) and P. aeruginosa K767 ΔrpoS (RpoS-) strains using real-time quantitative PCR. Expression was normalized to rpoD and is reported relative to the untreated wild-type strain K767. Values are means ± standard deviation of the means (STDEV) (error bars) from at least three independent determinations, each performed in triplicate. 50 positively affects aminoglycoside resistance was ruled out. To assess the second hypothesis that PA2797-PA2798 negatively affects RpoS activity with RpoS negatively affecting aminoglycoside resistance (i.e. loss of PA2797-PA2798 increases RpoS activity which reduces aminoglycoside resistance), the rpoS gene was cloned in a multicopy number plasmids (pRK415, a low copy number plasmid and pUCP19, a high copy number plasmid) and introduced in to P. aeruginosa K767. If the second hypothesis was true, then having an excess amount of rpoS in the bacterial cell would result in a decrease in aminoglycoside resistance. As shown in Table 3.2 there was no change in aminoglycoside resistance level between P. aeruginosa K767 carrying the rpoS-expressing plasmids and P. aeruginosa K767 carrying the empty plasmids. Based on this result, an excess of rpoS in P. aeruginosa K767 strain does not decrease aminoglycoside resistance. If the hypothesis was true and PA2797-PA2798 has a negative effect on RpoS, then their presence in P. aeruginosa K767 negatively impacts RpoS and does not allow for its true impact on aminoglycoside resistance. To avoid this issue, PA2797 and PA2798 were knocked out in P. aeruginosa K767 carrying empty plasmids, and plasmids expressing rpoS gene. If the hypothesis was true and PA2797-PA2798 negatively affects RpoS activity and, thus, impacts the amount of functional RpoS being produced, then their elimination should permit the plasmid-expressed RpoS to negatively impact aminoglycoside resistance. P. aeruginosa K767 carrying the plasmids expressing rpoS and lacking either PA2797 or PA2798 was subjected to antimicrobial susceptibility assays. As shown in Tables 3.3 and 3.4, there was no change in aminoglycoside resistance in rpoS-expressing P. aeruginosa K767 lacking PA2797 or PA2798. Thus, multi-copy rpoS does not decrease aminoglycoside resistance regardless of the presence or absence of PA2797-PA2798. Still, it was possible that RpoS is regulated post-transcriptionally as it is in E. coli. The amount of functional 51 Table 3.2 Impact of cloned rpoS on aminoglycoside resistance in K767 Minimum Inhibitory Concentration (µg/ml)a Plasmid PAR SPC KAN STR AMI TOB pRK415 256 1024 128 32 4 1 pRK415::rpoS 256 1024 128 32 4 1 pUCP19 256 1024 128 32 4 1 pUCP19::rpoS 256 1024 128 32 4 1 a abbreviations used: PAR, paromomycin; SPC, spectinomycin; KAN, kanamycin; STR, streptomycin; AMI, amikacin; TOB, tobramycin 52 Table 3.3 Impact of cloned rpoS on aminoglycoside resistance in K767 ΔPA2797 Minimum Inhibitory Concentration (µg/ml)a Plasmid PAR SPC KAN STR AMI TOB pRK415 16 256 16 8 0.5 0.125 pRK415::rpoS 16 256 16 8 0.5 0.125 pUCP19 16 256 16 8 0.5 0.125 pUCP19::rpoS 16 256 16 8 0.5 0.125 a abbreviations used: PAR, paromomycin; SPC, spectinomycin; KAN, kanamycin; STR, streptomycin; AMI, amikacin; TOB, tobramycin 53 Table 3.4 Impact of cloned rpoS on aminoglycoside resistance in K767 ΔPA2798 Minimum Inhibitory Concentration (µg/ml)a Plasmid PAR SPC KAN STR AMI TOB pRK415 16 256 16 8 0.5 0.125 pRK415::rpoS 16 256 16 8 0.5 0.125 pUCP19 16 256 16 8 0.5 0.125 pUCP19::rpoS 16 256 16 8 0.5 0.125 a abbreviations used: PAR, paromomycin; SPC, spectinomycin; KAN, kanamycin; STR, streptomycin; AMI, amikacin; TOB, tobramycin 54 RpoS in E. coli is controlled by ClpXP protease. Once RssB, an anti-RpoS anti-sigma factor is phosphorylated, it binds to RpoS; this complex is then recognized by ClpXP protease, which unfolds and degrades RpoS (Hengge-Aronis, 2002). Thus, although cloning rpoS in multi-copy plasmids might increase rpoS transcript levels, there may be no increase in active RpoS due to RpoS post-transcriptional regulation. As such, attempts to test hypothesis 2 by overexpressing rpoS may be fruitless. Another approach to test the hypothesis that PA2797-PA2798 negatively affects RpoS activity with RpoS negatively affecting aminoglycoside resistance (i.e. loss of PA2798 increases RpoS activity which reduces aminoglycoside resistance), which avoids posttranscriptional regulation of RpoS, is to knock out rpoS in P. aeruginosa K767 ΔPA2797 and P. aeruginosa K767 ΔPA2798. If loss of PA2797-PA2798 is negatively impacting aminoglycoside resistance by increasing RpoS activity, loss of rpoS in PA2797-PA2798 mutants should restore aminoglycoside resistance. An in-frame deletion of rpoS in P. aeruginosa K767 ΔPA2798 was successful. ΔrpoS derivative of P. aeruginosa K767 ΔPA2797 was not recorded despite several attempts. As shown in Table 3.5, loss of rpoS in P. aeruginosa K767 ΔPA2798 strain K3162 had no impact on aminoglycoside resistance, with the exception of a modest 2- fold decrease in paromomycin, streptomycin and tobramycin resistance level. While the nature of the decrease is unknown, it does not support the hypothesis. Based on these results, it is concluded that PA2797PA2798 is not connected to P. aeruginosa resistance through RpoS. 3.2 The connection between Anr and P. aeruginosa aminoglycoside resistance Microarray data for P. aeruginosa K767 ΔPA2798 revealed an increase in the expression of genes involved in anaerobic respiration (eg. norBC) and regulated by an anaerobic regulator called Anr (K. Poole, unpublished). P. aeruginosa is an aerobic pathogen, but it can also live in an environment where there is a lack of O2 (i.e. anaerobic environment). In an anaerobic 55 Table 3.5 Impact of loss of rpoS on aminoglycoside resistance in K767 ΔPA2798 Strain PA2798a rpoSb Minimum Inhibitory Concentration (µg/ml)c PAR SPC KAN STR AMI TOB K3162 - + 32 256 16 8/16 0.5/1 0.25/0.5 K3627 - - 16 256 16 4 0.5 0.125 a -, ΔPA2798. b +, wild-type rpoS; -, ΔrpoS. c abbreviations used: PAR, paromomycin; SPC, spectinomycin; KAN, kanamycin; STR, streptomycin; AMI, amikacin; TOB, tobramycin 56 environment nitrite, nitrate, and nitrous oxide are used in the place of O2 as alternative electron acceptors of the electron transport chain pathway (Zumft, 1997) and Anr plays a key role in activating this pathway (Trunk et al., 2010). Aminoglycosides enter the bacterial cell via an energy-dependent process (Taber et al., 1987). Although the exact mechanism by which aminoglycoside uptake occurs is still not defined, there is a common knowledge that this antibiotic relies on the energy provided by the electron transport chain and the respiratory pathway to enter the cell and reach its target, the ribosome (Bryan and Van Den Elzen, 1977). Thus, alterations in Anr-regulated respiration in the P. aeruginosa K767 ΔPA2798 strain might possibly impact aminoglycoside uptake and, thus, decrease aminoglycoside resistance. To test the hypothesis that PA2797-PA2798 negatively impacts Anr, with its loss upregulating Anr and enhancing aminoglycoside susceptibility, we first validated the array data by examining expression of the Anr target gene, norB, in the P. aeruginosa K767 ΔPA2798 mutant and compared it to wild-type P. aeruginosa K767. If the microarray result was accurate, then expression of the norB gene in P. aeruginosa K767 ΔPA2798 would increase relative to the parent strain P. aeruginosa K767. As shown in Figure 3.2, a 2-fold increase in norB expression was seen in P. aeruginosa K767 ΔPA2798.This increase in norB expression is consistent with the initial microarray result and confirms that Anr-regulated gene expression increases in P. aeruginosa K767 ΔPA2798. To assess whether an increase in expression of Anr- regulated genes in the P. aeruginosa K767 ΔPA2798 mutant was responsible for decrease in aminoglycoside resistance, the anr gene was deleted in P. aeruginosa K767 lacking either PA2797 or PA2798. Impact on aminoglycoside resistance was assessed. If the above was true, then knocking out the anr gene in P. aeruginosa K767 ΔPA2797 or ΔPA2798 would restore aminoglycoside resistance to wild- type levels. While an anr deletion was successfully introduced in P. aeruginosa K767 57 norB expression (fold-change) 1.4 1.2 1 0.8 0.6 0.4 0.2 0 PA2798+ PA2798- Figure 3.2 Impact of loss of PA2798 on norB expression in P. aeruginosa. The expression of norB was assessed in wild-type P. aeruginosa K767 strain (PA2798+) and P. aeruginosa K767 ΔPA2798 (PA2798-) strains using real-time quantitative PCR. Expression was normalized to rpoD and is reported relative to the untreated wild-type strain K767. Values are means ± STDEV from at least three independent determinations, each performed in triplicate. 58 ΔPA2798, Δanr derivatives of P. aeruginosa K767 ΔPA2797 was not obtained, despite several attempts. As shown in Table 3.6, no change in aminoglycoside resistance was observed in P. aeruginosa K767 Δ PA2798 regardless of the presence/ absence of anr, with the exception of a modest 2- fold increase in spectinomycin and tobramycin resistance in the strain lacking anr. To make sure that Δanr strain truly lacked the expression of anr, the expression of norB, an Anr target gene, was assessed using real-time PCR. In contrast to Anr+ strain, the Anr – strain showed no expression of norB. This result also showed that Anr was the sole regulator of norB gene and an increase in norB expression in P. aeruginosa K767 lacking PA2798, as previously showed in the microarray data as well as the real-time PCR, was due solely to the increase in Anr activity in this mutant strain. The above results do not support a link between PA297-PA2798 and aminoglycoside resistance in P. aeruginosa through Anr. A second approach was designed to test the hypothesis that an increase in expression of Anr-regulated genes in the P. aeruginosa K767 ΔPA2798 mutant was responsible for a decrease in aminoglycoside resistance. The second attempt was expressing anr from a multi-copy plasmid in wild-type strain K767 and assessing whether this enhanced susceptibility. As shown in Table 3.7, no change in aminoglycoside resistance was observed in P. aeruginosa K767 carrying anr plasmid relative to vector control strain. This experiment also failed to show a connection between Anr and aminoglycoside resistance. Based on these results, it is concluded that PA2797-PA2798 is not connected to P. aeruginosa resistance through Anr. 3.3 The connection between oxidative stress and P. aeruginosa aminoglycoside resistance Microarray results from P. aeruginosa K767 ΔPA2798 also showed an up-regulation of genes involved in the oxidative stress response, including lex A and recN, encoding proteins involved in DNA repair during the oxidative stress response process (Schlacher et al., 2006). 59 Table 3.6 Impact of loss of anr on aminoglycoside resistance in K767 ΔPA2798 Strain PA2798a anrb Minimum Inhibitory Concentration (µg/ml)c PAR SPC KAN STR AMI TOB K3162 - + 32 256 16 8/16 0.5/1 0.25/0.5 K3628 - - 32 512 16 8/16 0.5/1 0.125 a -, ΔPA2798. b +, wild-type anr; -, Δanr c abbreviations used: PAR, paromomycin; SPC, spectinomycin; KAN, kanamycin; STR, streptomycin; AMI, amikacin; TOB, tobramycin 60 Table 3.7 Impact of cloned anr on aminoglycoside resistance in K767 Minimum Inhibitory Concentration (µg/ml)a Plasmid PAR SPC KAN STR AMI TOB pRK415 256 512 64 16 2 1 pRK415::anr 256 512 64 32 2 1 128/256 512 64 32 2 1 64 512 32 32 2 1 pUCP19 pUCP19::anr a abbreviations used: PAR, paromomycin; SPC, spectinomycin; KAN, kanamycin; STR, streptomycin; AMI, amikacin; TOB, tobramycin 61 Organisms that grow aerobically are constantly faced with the generation of ROS as by-products of aerobic respiration. ROS are lethal to the bacterial cell, damaging proteins, membrane, lipids, as well as DNA (Dwyer et al., 2009). Therefore, exposure to ROS activates a variety of oxidative stress response pathways by which the organism tries to overcome the lethal effects of ROS. It is also known that exposure to a variety of antibiotics, including aminoglycosides, can generate ROS in bacterial cells. One possible effect of ROS generated due to the bacteria exposure to aminoglycoside might be membrane perturbation which in turn might increase aminoglycoside uptake (Kohanski et al, 2000), and, thus, decrease aminoglycoside resistance. Considering that oxidative stress partly explains aminoglycoside toxicity, then the increase in oxidative stress (as shown by the increase in the expression of lexA and recN) might contribute to the increase in aminoglycoside susceptibility in P. aeruginosa K767 ΔPA2798. Therefore, it was hypothesized that PA2797-PA2798 might be linked to aminoglycoside resistance through the organisms’ response to aminoglycoside-generated ROS, with loss of PA2797-PA2798 leading to an increase in ROS and a decrease in aminoglycoside resistance. To verify the results acquired from the microarray data, the expression of lexA and recN genes were assessed in P. aeruginosa K767 and P. aeruginosa K767 ΔPA2798, using real-time PCR. As shown in Figures 3.3, no increase in lexA or recN expression was seen in P. aeruginosa K767 ΔPA2798 relative to P. aeruginosa K767. Since the array data could not be validated, a link between the oxidative stress response and aminoglycoside resistance was not pursued. 3.4 The connection between temperature stress and P. aeruginosa aminoglycoside resistance Examination of the array data from P. aeruginosa K767 ΔPA2798 also revealed a downregulation of several genes involved in the heat shock response, including grpE, htpG, 62 2 1.8 1.6 Fold-change 1.4 1.2 1 recN 0.8 lexA 0.6 0.4 0.2 0 ΔPA2798 WT Figure 3.3 Impact of loss of PA2798 on lexA and recN expression in P. aeruginosa. The expression of lexA and recN was assessed in wild-type P. aeruginosa K767 strain (WT) and P. aeruginosa K767ΔPA2798 (ΔPA2798) strains using real-time quantitative PCR. Expression was normalized to rpoD and is reported relative to the untreated wild-type strain K767. Values are means ± STDEV from at least three independent determinations, each performed in triplicate. 63 and dnaJ. Bacteria live in environments in which temperature is optimal for their growth. In temperatures higher than the optimal level, proteins become misfolded. Heat shock related genes become induced in this situation in an attempt to repair misfolded proteins by either refolding them to the right structure, or help their degradation by transferring the misfolded proteins to proteases (Morimoto et al., 1997; Goldberg, 2003). Heat shock response genes are also induced in the presence of aminoglycosides (Kindrachuk et al., 2011). Kindrachuk et al. in 2011 showed that exposure to aminoglycosides increased expression of heat shock genes in P. aeruginosa. As mentioned previously, once aminoglycosides enter the bacterial cell, they attach to the ribosome, the aminoglycoside target site, and cause mistranslation and misfolding of proteins, such as membrane proteins (Davis et al., 1986; Taber et al., 1987). Presumably, heat shock genes are expressed in this situation to repair the misfolded proteins or help with their degradation, alleviating their deleterious effects on cell. Consistent with this, P. aeruginosa with a deficient rpoH gene, encoding the heat shock response sigma factor, showed a 2-fold decrease in aminoglycoside resistance (Kindrachuk et al., 2011). Thus, the increase in aminoglycoside susceptibility in P. aeruginosa K767 ΔPA2798 may result from a decrease in heat shock gene expression and loss of their protective effect. Because many heat shock genes are essential (Fayet et al., 1989) and previous attempt to construct an rpoH knock out was unsuccessful (C. Lau, unpublished), an initial effort of assessing a link between heat shock and aminoglycoside resistance via PA2797-PA2798 involved examining the impact of loss of PA2797-PA2798 on growth of P. aeruginosa in elevated temperatures. The expectation was that if the decrease in heat shock gene expression was sufficient to impact aminoglycoside resistance, it would also impact growth in high temperatures. 64 As shown by growth curves in Figure 3.4, while loss of PA2797-PA2798 compromised growth relative to wild-type P. aeruginosa K767, this was seen at all temperatures and generalized growth defect and was not related to growth temperature. Thus, PA2797-PA2798 does not appear to impact aminoglycoside resistance via heat shock response. 3.5 The connection between ribosomal genes and P. aeruginosa aminoglycoside resistance Microarray result of P. aeruginosa K767 Δ PA2798 showed down-regulation of several ribosomal genes, including a ribosomal protein operon comprised of rpsP (which encodes 30S ribosomal protein 16S), trmD (which encodes tRNA-(guanine-N1)-methyl transferase), rimM (which is involved in rRNA processing), and rplS (which encodes 50S ribosomal protein L19). Since aminoglycosides target the ribosome and ribosomal mutations are known to promote aminoglycoside resistance (Jana and Deb, 2006), it is not unexpected for change in ribosomal gene expression to impact aminoglycoside resistance. To assess whether the decrease in expression of the 4-gene ribosomal operon in P. aeruginosa K767 ΔPA2798 was responsible for the increase in aminoglycoside susceptibility of this strain, array data was first validated by determining whether the operon expression decreased in P. aeruginosa K767 ΔPA2798 strain, using rpsP as a representative gene. As shown in Figure 3.5, q-RT revealed a 2-4 fold decrease in operon expression in P. aeruginosa K767 ΔPA2798 as compared to P. aeruginosa K767. Having confirmed the microarray data, it was then assessed whether a decrease in ribosomal gene expression was linked to the decrease in aminoglycoside resistance in P. aeruginosa K767 ΔPA2798. To do this, an attempt to restore expression of the ribosomal operon in P. aeruginosa K767 ΔPA2798 was made by cloning it on multi-copy vectors and then assess the impact on aminoglycoside resistance. rpsP operon was cloned in plasmids pRK415 (low copy number) and 65 Growth (OD600) 2 A 1.5 K767 1 Δ2797 0.5 Δ2798 0 0 2 4 6 Time(hrs) Growth (OD600) 2 B 1.5 1 K767 Δ2797 0.5 Δ2798 0 0 2 4 6 Time(hrs) Growth (OD600) 2 C 1.5 1 K767 0.5 Δ2797 Δ2798 0 0 2 4 6 Time(hrs) Figure 3.4 Impact of loss of PA2797-PA2798 on growth of P. aeruginosa. P. aeruginosa strain K767, K767 ΔPA2797 and K767 ΔPA2798 were incubated at A) 30°C, B) 37°C and C) 42°C and growth was assessed over time. Results are representative of independent experiments. 66 rpsP expression (fold-change) 1.2 1 0.8 0.6 0.4 0.2 0 PA2798+ PA2798- Figure 3.5 Impact of loss of PA2798 on rpsP expression in P. aeruginosa K767. The expression of rpsP was assessed in wild-type P. aeruginosa K767 strain (PA2798+) and P. aeruginosa K767 ΔPA2798 (PA2798-) using real-time quantitative PCR. Expression was normalized to rpoD and is reported relative to the untreated wild-type strain K767. Values are means ± STDEV from at least three independent determinations, each performed in triplicate 67 pUCP19 (high copy number) and was introduced in to P. aeruginosa K767 ΔPA2798 and aminoglycoside resistance was assessed. As shown in Table 3.8, there was no change in aminoglycoside resistance of P. aeruginosa K767 ΔPA2798 carrying pRK415::rpsP operon or pUCP19::rpsP operon as compared to empty vector controls. To ensure that enhanced expression of the ribosomal operon had occurred in the strain harboring the rpsP operon- containing plasmids, the expression of rpsP (as a representative of the operon) was assessed in plasmidbearing P. aeruginosa K767 ΔPA2798. As shown in Figure 3.6, there was an approximate 8-fold change increase in the expression of rpsP in P. aeruginosa K767 ΔPA2798 carrying the rpsP operon plasmid as compared to the empty vector control. This result indicated that expression of rpsP operon was restored. Since there was no increase in aminoglycoside resistance in P. aeruginosa K767 ΔPA2798 upon restoration of operon expression, it was concluded that the increase in aminoglycoside susceptibility of the P. eruginosa K767 ΔPA2798 mutant was not directly related to the decrease in expression of the rpsP operon. 68 Table 3.8 Impact of cloned rpsP operon on aminoglycoside resistance in K767 ΔPA2798 Minimum Inhibitory Concentration (µg/ml)a Plasmid PAR SPC KAN STR AMI TOB pRK415 32 256 16 8 0.5 0.125 pRK415::rpsP operon 32 256 16 8 0.5 0.125 pUCP19 16 256 16 8 0.5 0.25 pUCp19::rpsP operon 16 256 16 8 0.5 0.25 a abbreviations used: PAR, paromomycin; SPC, spectinomycin; KAN, kanamycin; STR, streptomycin; AMI, amikacin; TOB, tobramycin 69 rpsP expression (fold-change) 1.2 1 0.8 0.6 0.4 0.2 0 pRK415 pRK415::rpsP operon Figure 3.6 Expression of rpsP as a measure of rpsP operon expression in P. aeruginosa K767 ΔPA2798. Expression of rpsP was assessed in P. aeruginosa strain K767 ΔPA2798 carrying the plasmid pRK415 with the cloned rpsP operon using real-time quantitative PCR. Expression was normalized to rpoD and is reported relative to the untreated wild-type strain K767. Values are means ± STDEV from at least three independent determinations, each performed in triplicate. 70 Discussion Aminoglycosides are used as one of the most common antibiotics in the treatment of pulmonary infections, caused by P. aeruginosa in patients suffering from CF (Cheer et al., 2003). P. aeruginosa is, however, becoming more resistant to aminoglycosides, compromising aminoglycosides efficacy in treating the infections caused by this microorganism (MacLeod et al., 2000; Shawar et al., 1999; Price et al., 1981; Saavedra et al., 1986). Therefore, identifying factors involved in P. aeruginosa resistance to aminoglycoside is vital and has been subject of much investigation. In 2012, Krahn et al. performed a random mutagenesis assay on a P. aeruginosa panaminoglycoside clinical isolate in search for genes that were intrinsically involved in aminoglycoside resistance. Transposon insertion in genes encoding an atypical regulatory system, PA2797-PA2798, produced a decrease in aminoglycoside resistance that was further confirmed by individually deleting the PA2797 and PA2798 genes. PA2797 is annotated as an anti-anti sigma factor which is inactivated by its phosphorylation by an unknown kinase; PA2798 encodes a probable sensor phosphatase (by Pseudomonas project; http://www.Pseudomonas.com). It has homology to E. coli RssB, an anti-sigma factor of a general stress response sigma factor, RpoS. PA2797 and PA2798 are also respective homologues of Bacillus SpoIIAA and SpoIIE proteins (Errington et al., 1996) as well as Bacillus RsbV and RsbU proteins (Hecker et al., 2007). SpoIIAA and SpoIIE regulate the SpoIIAB anti-sigma factor of the sporulation sigma factor σF, while RsbV and RsbU regulate the RsbW anti-sigma factor of the general stress response sigma factor σB. Since PA2797-PA2798 are respective homologues of Bacillus SpoIIAA and SpoIIE proteins as well as Bacillus RsbV and RsbU 71 proteins, it is possible that PA2797-PA2798 are also involved in the regulation of a sigma factor in P. aeruginosa. In Bacillus subtilis major changes in the pattern of transcription during sporulation are brought about by the synthesis of new sigma factors, including σF. Sigma F is negatively regulated by an anti-sigma factor SpoIIAB (Duncan and Losick, 1993; Min et al., 1993), which is in turn controlled by an anti-anti-sigma factor SpoIIAA (Partridge et al., 1991; Schmidt et al., 1990). spoIIAA and spoIIAB along with spoIIAC, encoding σF, are coordinately regulated in an operon called spoIIA operon (Figure 4.1A). In the presence of ATP, SpoIIAA is inactivated by phosphorylation on a specific serine residue. SpoIIAA then remains inactive, allowing SpoIIAB to inhibit σF. In the presence of ADP however, the active SpoIIAA binds to SpoIIAB, preventing it from binding to σF (Errington et al., 1996). A fourth protein SpoIIE though not residing on the same operon, also acts in favor of σF by either independently acting as SpoIIAB antagonist or by acting as a phosphatase to regenerate an active, non-phosphorylated form of SpoIIAA (Partridge et al., 1991; Margolis et al., 1991) The regulation of B. subtilis general stress response sigma factor, σB, is similar to the regulation of B. subtilis sporulation sigma factor, σF. Sigma B is negatively regulated by an antisigma factor RsbW which in turn is controlled by an ant-anti-sigma factor RsbV (Dufour and Haldenwang, 1994). rsbV, rsbW, and σB are all part of sigma B operon (Figure 4.1B). In growing cells, RsbV is phosphorylated by RsbW, and therefore, it is inactive, allowing a tight attachment between RsbW and σB, leading to the inactivation of σB. In response to a presence of stress, nonphosphorylated RsbV, generated by either of two RsbV∼P-specific phosphatases, RsbU and RsbP, binds to RsbW, releasing σB from the inhibitory RsbW/Sigma B complex (Vijay et al., 2000; Yang et al., 1996). 72 Figure 4.1 The regulation of σF and σB in B. subtilis. A) Sigma F is negatively regulated by an anti-sigma factor SpoIIAB, which is in turn controlled by an anti-anti-sigma factor SpoIIAA. SpoIIAA is activated in the presence of ADP, binding to SpoIIAB, releasing σF. In the presence of ATP, SpoIIAA is inactivated, allowing SpoIIAB to bind tightly to σF, repressing its function. SpoIIE acts either as SpoIIAB antagonist or SpoIIAA phosphatase, regenerating active SpoIIAA. Possible place of PA2797 and PA2798 in a P. aeruginosa sigma factor regulating pathway, similar to the B. subtilis σF regulating pathway model is shown in the figure. B) Sigma B is negatively regulated by an anti-sigma factor RsbW which in turn is controlled by an ant-anti-sigma factor RsbV. Inactive RsbV is generated by its phosphorylation by RsbW. RsbW can then bind tightly to σB and inactivate it. In response to a presence of stress, nonphosphorylated RsbV, generated by either of two RsbV∼P-specific phosphatases, RsbU and RsbP, binds to RsbW, releasing σB from the inhibitory RsbW/Sigma B complex. Possible place of PA2797 and PA2798 in a P. aeruginosa sigma factor regulating pathway, similar to the B. subtilis σB regulating pathway model is shown in the figure. 73 A) SpoIIE σF SpoIIAA SpoIIAB spoIIE spoIIAA PA2798 spoIIAC spoIIAB PA2797 B) rsbRA σA rsbS rsbT RsbU RsbV RsbW σB rsbU rsbV rbsW sigB PA2798 σ rsbX RsbP PA2797 B 74 rsbQ rsbP The aim of this study was to identify possible pathways that connected PA2797-PA2798 to P. aeruginosa aminoglycoside resistance, and the previous microarray analysis (K. Poole, unpublished) was used as an initial guide to further identify genes that possibly had an impact on PA2797-PA2798, and aminoglycoside resistance relation. Genes whose connection was investigated in this study were rpoS, anr, oxidative stress related genes, heat shock-related genes, and a ribosomal protein operon (rpsP operon). A possible connection between RpoS and PA2797-PA2798 was investigated due to PA2798 structural similarity to RssB, an E. coli antisigma factor of RpoS (Hengge-Aronis, 2002). Also, since P. aeruginosa PA2797-PA2798 are homologous of Bacillus regulators of different sigma factors, such as δF (Errington et al., 1996) and δB (Min et al., 1993), it was reasonable to investigate whether PA2797-PA2798 regulated the RpoS sigma factor. A series of experiments, including gene deletion, gene cloning, and q-RT PCR was performed. No problem was encountered during the investigation process. The only issue present was an unsuccessful deletion of rpoS gene in P. aeruginosa K767 ΔPA2797. Although the mating process was successful, no strain with PA2797 and rpoS double deletion was detected by screening through colony PCR. It is possible that constructing an additional knock out in K767 ΔPA2797 cause self-synthetic lethality for unknown reasons; therefore, it is not possible to make rpoS knock out in this mutant strain. Based on the given results, there is no apparent connection between RpoS, PA2797-PA2798 and aminoglycoside resistance in P. aeruginosa. The next pathway being investigated was the involvement of Anr in P. aeruginosa aminoglycoside resistance. Anr was chosen for further investigation due to its possible role in the amount of energy provided for the uptake of aminoglycosides. As mentioned previously, aminoglycosides depend on the energy provided by the respiration pathway and electron 75 transport chain to enter the cell (Bryan et al., 1979; Bryan and Kwan, 1983). If the amount of energy provided by these pathways changes it probably impacts the amount of aminoglycoside entering the cell in both EDPI and EDPII stages, thus changing the aminoglycosides efficacy on the bacterial cell. The microarray assay on P. aeruginosa K767 ΔPA2798 showed an upregulation of the genes regulated by Anr, an anaerobic pathway main regulator. Since the expression of the genes involved in an anaerobic pathway was up-regulated, it was possible that a respiratory pathway regulated by Anr was activated even though the environment was aerobic. In other words, deletion of PA2798 may have mimicked a condition in which the respiration pathway regulated by Anr is normally activated. This change in respiratory pathway may impact the amount of energy provided by the electron transport chain and the respiratory pathway, thus affecting the amount of aminoglycoside entering the bacterial cell. Therefore, decrease in aminoglycoside resistance level in P. aeruginosa K767 ΔPA2798 may be due to the involvement of Anr. A series of experiments, including gene deletion, gene cloning, and q-RT PCR was performed. No problem was encountered during the investigation process. The only issue present was an unsuccessful deletion of anr gene in P. aeruginosa K767 ΔPA2797. Although the mating process was successful, no strain with PA2797 and anr double deletion was detected by screening through colony PCR. It is possible that constructing an additional knock out in K767 ΔPA2797 cause self-synthetic lethality for unknown reasons; therefore, it is not possible to make anr knock out in this mutant strain. Based on the results gathered from the experiments, there is no detected connection between Anr, PA2797-PA2798, and aminoglycoside resistance in P. aeruginosa. The next step was to investigate the possible involvement of oxidative stress in P. aeruginosa aminoglycoside resistance. Among genes whose expression was changed in P. aeruginosa K767 76 ΔPA2798, the microarray data showed an up-regulation of genes such as lexA and recN that are involved in oxidative stress response. As mentioned previously, one of the possible impacts of aminoglycosides on the bacterial cells is the generation of ROS which damages the bacterial membrane and increases the amount of aminoglycoside uptake in EDPII (Kohanski et al., 2007). ROS create oxidative stress in the bacterial cell, damaging molecules such as DNA, lipids, and proteins. Bacteria activate an oxidative stress response mechanism to overcome the lethal effects of ROS. Up-regulation of genes involved in oxidative stress response mechanism in P. aeruginosa K767 ΔPA2798 may be then due to an increase in the level of oxidative stress in this strain which may be due to more uptake of aminoglycosides. Decrease in aminoglycoside resistance level in P. aeruginosa K767 ΔPA2798 might be then explained by the connection between oxidative stress and PA2797-PA2798. Therefore, a possible connection between oxidative stress and PA2797-PA2798 was investigated. Despite several attempts, the microarray result could not be confirmed using the real-time PCR. The q-RT result was inconsistent though conditions at which RNAs were prepared, extracted, and converted to cDNA were optimum. The condition at which q-RT PCR was performed for each gene of interest was also previously optimized and the experiment’s accuracy was confirmed using a reference gene rpoD. Since no solid result could be gathered from the q-RT PCR experiment, further investigation on a possible connection between oxidative stress and PA2797-PA2798 was not performed. The possible impact of heat shock-related genes on the PA2797-PA2798 and aminoglycoside resistance relation was next investigated since there was a decrease in the expression of genes involved in heat shock response in P. aeruginosa K767 strain lacking PA2798, as well as previously confirmed involvements of heat shock-related genes in aminoglycoside resistance in P. aeruginosa by Kindrachuk in 2012. Kindrachuk et al. (2012) showed that in a P. aeruginosa 77 strain carrying a mutation in rpoH, a heat shock stress response sigma factor, there was a modest 2-fold decrease in aminoglycoside resistance. A microarray assay performed by Poole lab (K. Poole, unpublished) showed reduced expression of heat shock-related genes, such as hscB, dnaJ, and htpG in P. aeruginosa K767 lacking PA2798. Therefore, it was hypothesized that the decrease in aminoglycoside resistance in this mutant might be connected to a reduced expression of heat shock- regulated genes. Despite several attempts in the Poole lab in the past, the deletion of rpoH in P. aeruginosa K767 has never been achieved. Still, if the P. aeruginosa K767 ΔPA2798 mutant had a defect in the heat shock response and this was causing an increase in aminoglycoside susceptibility, a defect in growth at elevated temperatures should have been seen. A growth rate experiment was performed; however, the result didn’t show any connection between the heat shock stress response, and PA2797-PA2798. One possibility as to why no connection was detected between heat shock stress response and PA2797-PA2798, is that it might be possible that the effect of rpoH on PA2797-PA2798 is modest; therefore, it does not impact growth at elevated temperatures. The last step was to investigate the possible involvement of the rpsP ribosomal protein operon in aminoglycoside resistance. Since the ribosome is the aminoglycosides target site (Jana and Deb, 2006) any change in the structure or function of the ribosome might affect the efficacy of the aminoglycosides. The microarray result showed down-regulation of some of the ribosomal genes, including this particular ribosomal protein operon, rpsP operon, in P. aeruginosa K767 ΔPA2798; therefore, possible involvement of the operon in aminoglycoside resistance was further investigated. q-RT PCR confirmed the microarray result, allowing the further experiments to search for the possible connection between the ribosomal protein operon and aminoglycoside resistance. Since the operon contained two essential genes (rpsP and rplS), it 78 was not possible to knock out the entire operon, so the operon was rather cloned in to multi-copy vectors and its effect on aminoglycoside resistance was investigated. No problem was encountered during the experiments performed; however, the final results showed no connection between the ribosomal operon, PA2797-PA2798, and aminoglycosides resistance. One possibility as to why no connection between ribosomal genes and aminoglycoside resistance was detected might be that there were several other ribosomal genes whose expression was also down-regulated in P. aeruginosa K767 ΔPA2798, and these genes along with genes residing on rpsP operon may have collectively contributed to the decrease in aminoglycoside resistance in this mutant strain. Therefore, restoring the expression of only some of these ribosomal genes is insufficient to restore aminoglycoside resistance in P. aeruginosa K767 ΔPA2798. There are possible explanations as to why the pathway by which PA2797-PA2798 and aminoglycoside resistance are connected was not identified in this study. PA2797-PA2798 as a two component system is involved in a regulation of a variety of pathways, which may be connected. Therefore, it is possible that deactivating or restoring the function of only one pathway at a time does not affect PA2797-PA2798. It is possible that multiple pathways collectively affect PA2797-PA2798 function, and since in this study these possible pathways were studied individually, no satisfying results were obtained. This study is based on the initial results from the microarray analysis performed previously (K. Poole, unpublished data). It is possible that the target gene was not identified in the array due to the microarray analysis common errors (Weng et al., 2006; Rocke, D.M. and Durbin, B. 2001). Another possibility is that the target gene was in fact identified in the microarray analysis, but was not chosen for further investigation. There were many genes whose expression was changed 79 in the mutant strain P. aeruginosa K767 with an in-frame deletion of PA2798, but it was not feasible to do an experiment on every gene detected in the microarray assay. 80 Conclusion This study aimed to identify pathways by which the putative two component system PA2797PA2798 is connected to P. aeruginosa aminoglycosides. A possible connection between PA2797-PA2798 and RpoS, a general stress regulator, Anr, an anaerobic respiration regulator, oxidative stress response, temperature stress response, and ribosomal proteins was investigated, but no connection was detected. However, there are still possible places for further investigation. All the steps from aminoglycoside uptake to the metabolic processes disrupted by aminoglycosides can be a potential place of investigation. Any alterations in pathways that provide energy for aminoglycoside uptake, electrochemical potential across the cytoplasm as well as the electron flow through the membrane bound respiratory chain, can affect aminoglycosides efficacy since EDPI and EDPII phases of aminoglycoside uptake are energy-dependent and this energy is provided by the electron transport chain and the membrane potential. Experiments performed by El’Garch et al. in 2007 and Fajardo et al. in 2008 have already shown that the membrane potential, and the electron transport chain, can also contribute to aminoglycoside resistance in P. aeruginosa. The initial microarray analysis on P. aeruginosa K767 ΔPA2798 (K. Poole, unpublished) showed an increase in the expression of genes whose products were involved in the electron transport chain and the membrane potential, including narK1, encoding a membrane protein, PA0567, encoding the membrane potential modulator, PA0521, encoding a protein involved in oxidation-reduction process, and nirN, encoding a c-type cytochrome. These four genes and their possible involvement in PA2797-PA2798 and aminoglycoside resistance pathway can also be part of 81 future investigation since their contribution to the electron transport chain and the membrane potential might affect the amount of energy provided by these two systems for aminoglycoside uptake, and therefore, might affect aminoglycoside resistance. Any structural or functional alteration of the aminoglycoside target site, the ribosome, can change aminoglycoside level of resistance in P. aeruginosa. In 2007, El’Garch et al. identified a ribosomal gene, rplY, whose disruption resulted in increased resistance to aminoglycosides. The microarray analysis (K. Poole, unpublished) showed a decrease in the expression of ribosomal genes, including rplR, rplA, rplO, and rpsO, along with genes residing on rpsP ribosomal operon in P. aeruginosa K767 ΔPA2798. As mentioned in section 4, it might be possible that these ribosomal genes collectively contribute to aminoglycoside resistance in this mutant strain; therefore their involvement in aminoglycoside resistance can be a potential target for further investigation. Any mutation in genes whose products are involved in heat shock stress response, oxidative stress response, envelope stress response or any other stress responses being activated once the bacterial cell is exposed to aminoglycosides, can also be a potential place of further investigation. Demonstrated by Kohanski et al. (2007), Nguyen et al. (2011), Kindrachuk et al. (2011), and Krahn et al. (2012), mutations in genes whose products were involved in DNA damage-related stress response (SOS), stringent stress response, heat shock stress response, and envelope stress response contributed to aminoglycoside resistance. The microarray analysis of P. aeruginosa K767 ΔPA2798 (K. Poole, unpublished) also showed a decrease in stringent stress response- related gene (sspB), as well as heat shock- related genes (hscB, dnaJ, htpG), and an increase in oxidative stress-related genes (osmC, lexA, recN, recA), the involvement of some being investigated in this study. Further investigation on stress-related genes might also be a target of future studies. 82 References Aires, J. R., T. Köhler, H. Nikaido, and P. Plésiat, 1999: Involvement of an active efflux system in the natural resistance of Pseudomonas aeruginosa to aminoglycosides. Antimicrob. Agents Chemother., 43, 2624-2628. Almagro, P., M. Salvado, C. Garcia-Vidal, M. Rodriguez-Carballeira, E. Cuchi, J. Torres, and J. L. Heredia, 2012: Pseudomonas aeruginosa and mortality after hospital admission for chronic obstructive pulmonary disease. Respiration, 84, 36-43. Alvarez-Ortega, C., I. Wiegand, J. Olivares, R. Hancock, and J. L. Martínez, 2011: The intrinsic resistome of Pseudomonas aeruginosa to β-lactams. Virulence, 2, 144-146. Amsterdam, D., 1996: Susceptibility testing of antimicrobials in liquid media. Antibiotics in laboratory medicine, V. Lorian Ed., Williams & Wilkins, 52-111. Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. Seidman, J. A. Smith, and K. Struhl, 2002: Short protocols in molecular biology: a compendium of methods from current protocols in molecular biology. John Wiley & Sons, Inc., New York, N. Y. Beck, E., G. Ludwig, E. Auerswald, B. Reiss, and H. Schaller, 1982: Nucleotide sequence and exact localization of the neomycin phosphotransferase gene from transposon Tn5. Gene, 19, 327336. Bilgin, N., A. A. Richter, M. Ehrenberg, A. E. Dahlberg, and C. G. Kurland, 1990: Ribosomal RNA and protein mutants resistant to spectinomycin. EMBO J., 9, 735-739. Bonomo, R. A., D. Szabo, 2006: Mechanisms of multidrug resistance in Acinetobacter species and Pseudomonas aeruginosa. Clin. Infect. Dis., 43 (Suppl 2), S49-S56. Breidenstein, E., C. de la Fuente-Núñez, and R. E. Hancock, 2011: Pseudomonas aeruginosa: all roads lead to resistance. Trends Microbiol., 19, 419-426. Breidenstein, E. B., B. K. Khaira, I. Wiegand, J. Overhage, and R. E. Hancock, 2008: Complex 83 ciprofloxacin resistome revealed by screening a Pseudomonas aeruginosa mutant library for altered susceptibility. Antimicrob. Agents Chemother., 52, 4486-4491. Brodersen, D. E., W. M. Clemons Jr, A. P. Carter, B. T. Wimberly, and V. Ramakrishnan, 2002: Crystal structure of the 30S ribosomal subunit from Thermus thermophilus: structure of the proteins and their interactions with 16S RNA. J. Mol. Biol., 316, 725-768. Bryan, L., S. Kowand, and H. Van Den Elzen, 1979: Mechanism of aminoglycoside antibiotic resistance in anaerobic bacteria: Clostridium perfringens and Bacteroides fragilis. Antimicrob. Agents Chemother., 15, 7-13. Bryan, L., H. Van Den Elzen, 1977: Effects of membrane-energy mutations and cations on streptomycin and gentamicin accumulation by bacteria: a model for entry of streptomycin and gentamicin in susceptible and resistant bacteria. Antimicrob. Agents Chemother., 12, 163-177. Bryan, L. E., S. Kwan, 1983: Roles of ribosomal binding, membrane potential, and electron transport in bacterial uptake of streptomycin and gentamicin. Antimicrob. Agents Chemother., 23, 835-845. Burns, J. L., R. L. Gibson, S. McNamara, D. Yim, J. Emerson, M. Rosenfeld, P. Hiatt, K. McCoy, R. Castile, A. L. Smith, and B. W. Ramsey, 2001: Longitudinal assessment of Pseudomonas aeruginosa in young children with cystic fibrosis. J. Infect. Dis., 183, 444-452. Busch‐Sorensen, C., N. Frimodt‐Moller, G. H. Miller, and F. Espersen, 1996: Aminoglycoside resistance among Danish blood culture isolates of coagulase‐negative staphylococci. APMIS, 104, 873-880. Campbell, B. D., R. J. Kadner, 1980: Relation of aerobiosis and ionic strength to the uptake of dihydrostreptomycin in Escherichia coli. Biochim. Biophys. Acta, 593, 1-10. Carter, A. P., W. M. Clemons, D. E. Brodersen, R. J. Morgan-Warren, B. T. Wimberly, and V. Ramakrishnan, 2000: Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature, 407, 340-348. 84 Caughlan, R. E., S. Sriram, D. M. Daigle, A. L. Woods, J. Buco, R. L. Peterson, J. A. Dzink-Fox, S. Walker, and C. R. Dean, 2009: Fmt bypass in Pseudomonas aeruginosa causes induction of MexXY efflux pump expression. Antimicrob. Agents Chemother., 53, 5015-5021. Cheer, S. M., J. Waugh, and S. Noble, 2003: Inhaled Tobramycin (TOBI®). Drugs, 63, 25012520. Choi, K., A. Kumar, and H. P. Schweizer, 2006: A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: Application for DNA fragment transfer between chromosomes and plasmid transformation. J. Microbiol. Methods, 64, 391-397. Cundliffe, E., 1989: How antibiotic-producing organisms avoid suicide. Annu. Rev. Microbiol., 43, 207-233. Davies, J. C., D. Bilton, 2009: Bugs, biofilms, and resistance in cystic fibrosis. Respir. Care, 54, 628-640. Davis, B. D., 1987: Mechanism of bactericidal action of aminoglycosides. Microbiol. Rev., 51, 341-350. Davis, B. D., L. Chen, and P. C. Tai, 1986: Misread protein creates membrane channels: an essential step in the bactericidal action of aminoglycosides. Proc. Natl. Acad. Sci. U.S.A., 83, 6164-6168. Doi, Y., Y. Arakawa, 2007: 16S ribosomal RNA methylation: emerging resistance mechanism against aminoglycosides. Clin. Infect. Dis., 45, 88-94. Doi, Y., D. de Oliveira Garcia, J. Adams, and D. L. Paterson, 2007: Coproduction of novel 16S rRNA methylase RmtD and metallo-β-lactamase SPM-1 in a panresistant Pseudomonas aeruginosa isolate from Brazil. Antimicrob. Agents Chemother., 51, 852-856. Dornbusch, K., H. O. Hallander, 1980: Gentamicin resistance in Gram-negative bacilli: occurrence of modifying enzymes and their influence on susceptibility testing. Scand. J. Infect. Dis., 12, 295-302. 85 Dotsch, A., T. Becker, C. Pommerenke, Z. Magnowska, L. Jansch, and S. Haussler, 2009: Genomewide identification of genetic determinants of antimicrobial drug resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother., 53, 2522-2531. Driscoll, J. A., S. L. Brody, and M. H. Kollef, 2007: The epidemiology, pathogenesis and treatment of Pseudomonas aeruginosa infections. Drugs, 67, 351-368. Dufour, A., W. G. Haldenwang, 1994: Interactions between a Bacillus subtilis anti-sigma factor (RsbW) and its antagonist (RsbV). J. Bacteriol., 176, 1813-1820. Duncan, L., R. Losick, 1993: SpoIIAB is an anti-sigma factor that binds to and inhibits transcription by regulatory protein sigma F from Bacillus subtilis. Proc. Natl. Acad. Sci. U.S.A., 90, 2325-2329. Durante-Mangoni, E., A. Grammatikos, R. Utili, and M. E. Falagas, 2009: Do we still need the aminoglycosides? Int. J. Antimicrob. Agents, 33, 201-205. Dwyer, D. J., and Coauthors, 2014: Antibiotics induce redox-related physiological alterations as part of their lethality. Proc. Natl. Acad. Sci. U.S.A., 111, E2100-E2109. Dwyer, D. J., M. A. Kohanski, and J. J. Collins, 2009: Role of reactive oxygen species in antibiotic action and resistance. Curr. Opin. Microbiol., 12, 482-489. El'Garch, F., K. Jeannot, D. Hocquet, C. Llanes-Barakat, and P. Plésiat, 2007: Cumulative effects of several nonenzymatic mechanisms on the resistance of Pseudomonas aeruginosa to aminoglycosides. Antimicrob. Agents Chemother., 51, 1016-1021. Emerson, J., M. Rosenfeld, S. McNamara, B. Ramsey, and R. L. Gibson, 2002: Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr. Pulmonol., 34, 91-100. Errington, J., A. Feucht, P. J. Lewis, M. Lord, T. Magnin, S. M. Najafi, J. F. Wilkinson, and M. D. Yudkin, 1996: Control of the cell-specificity of sigma F activity in Bacillus subtilis. Phil. Trans. R. Soc. B, 351, 537-542. 86 Fajardo, A., N. Martínez-Martín, M. Mercadillo, J. C. Galán, B. Ghysels, S. Matthijs, P. Cornelis, L. Wiehlmann, B. Tümmler, and F. Baquero, 2008: The neglected intrinsic resistome of bacterial pathogens. PLoS One, 3, e1619. Fayet, O., T. Ziegelhoffer, and C. Georgopoulos, 1989: The groES and groEL heat shock gene products of Escherichia coli are essential for bacterial growth at all temperatures. J. Bacteriol., 171, 1379-1385. Fernández, L., E. Breidenstein, and R. E. Hancock, 2011: Creeping baselines and adaptive resistance to antibiotics. Drug Resist. Updat., 14, 1-21. Floret, N., X. Bertrand, M. Thouverez, and D. Talon, 2009: Nosocomial infections caused by Pseudomonas aeruginosa: Exogenous or endogenous origin of this bacterium? Pathol. Biol., 57, 9-12. Foweraker, J., 2009: Recent advances in the microbiology of respiratory tract infection in cystic fibrosis. Br. Med. Bull., 89, 93-110. Fraud, S., K. Poole, 2011: Oxidative stress induction of the MexXY multidrug efflux genes and promotion of aminoglycoside resistance development in Pseudomonas aeruginosa. Antimicrob. Agents Chemother., 55, 1068-1074. Freeman, S., J. C. Sharp, and M. Harrington, 2008: Biological science. 4th ed. BenjaminCummings, 1320pp. Galimand, M., T. Lambert, G. Gerbaud, and P. Courvalin, 1993: Characterization of the aac (6')Ib gene encoding an aminoglycoside 6'-N-acetyltransferase in Pseudomonas aeruginosa BM2656. Antimicrob. Agents Chemother., 37, 1456-1462. Gallagher, L. A., J. Shendure, and C. Manoil, 2011: Genome-scale identification of resistance functions in Pseudomonas aeruginosa using Tn-seq. mBio, 2, e00315-10. Gibson, R. L., J. L. Burns, and B. W. Ramsey, 2003: Pathophysiology and management of pulmonary infections in cystic fibrosis. Am. J. Respir. Crit. Care Med., 168, 918-951. 87 Goldberg, A. L., 2003: Protein degradation and protection against misfolded or damaged proteins. Nature, 426, 895-899. Govan, J. R., V. Deretic, 1996: Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol. Rev., 60, 539-574. Green and, R., H. F. Noller, 1997: Ribosomes and translation. Annu. Rev. Biochem., 66, 679-716. Griffith, L. J, 1966: Development of resistance to kanamycin. Ann. N. Y. Acad. Sci., 132, 796799. Griffith, L. J., W. E. Ostrander, and Z. F. Smith, 1960: A comparison of the in vitro effectiveness of kanamycin and five other antibacterial agents against common Gram-negative rod pathogens. Antibiot. Chemother., 10, 88–92 Gudiol, C., F. Tubau, L. Calatayud, C. Garcia-Vidal, M. Cisnal, I. Sanchez-Ortega, R. Duarte, M. Calvo, and J. Carratala, 2011: Bacteraemia due to multidrug-resistant Gram-negative bacilli in cancer patients: risk factors, antibiotic therapy and outcomes. J. Antimicrob. Chemother., 66, 657-663. Gurung, M., D. C. Moon, M. D. Tamang, J. Kim, Y. C. Lee, S. Y. Seol, D. T. Cho, and J. C. Lee, 2010: Emergence of 16S rRNA methylase gene armA and cocarriage of blaIMP-1 in Pseudomonas aeruginosa isolates from South Korea. Diagn. Microbiol. Infect. Dis., 68, 468-470. Hachler, H., P. Santanam, and F. H. Kayser, 1996: Sequence and characterization of a novel chromosomal aminoglycoside phosphotransferase gene, aph (3')-IIb, in Pseudomonas aeruginosa. Antimicrob. Agents Chemother., 40, 1254-1256. Halliwell, B., 2005: Free radicals and other reactive species in disease. John Wiley & Sons, Inc., New York, N. Y.. Hancock, R. E., V. J. Raffle, and T. I. Nicas, 1981: Involvement of the outer membrane in gentamicin and streptomycin uptake and killing in Pseudomonas aeruginosa. Antimicrob. Agents 88 Chemother., 19, 777-785. Hassett, D. J., M. D. Sutton, M. J. Schurr, A. B. Herr, C. C. Caldwell, and J. O. Matu, 2009: Pseudomonas aeruginosa hypoxic or anaerobic biofilm infections within cystic fibrosis airways. Trends Microbiol., 17, 130-138. Hatch, R. A., N. L. Schiller, 1998: Alginate lyase promotes diffusion of aminoglycosides through the extracellular polysaccharide of mucoid Pseudomonas aeruginosa. Antimicrob. Agents Chemother., 42, 974-977. Hay, T., S. Fraud, C. H. Lau, C. Gilmour, and K. Poole, 2013: Antibiotic inducibility of the mexXY multidrug efflux operon of Pseudomonas aeruginosa: involvement of the MexZ antirepressor ArmZ. PloS one, 8, e56858. Hecker, M., J. Pané-Farré, and V. Uwe, 2007: SigB-dependent general stress response in Bacillus subtilis and related Gram-positive bacteria. Annu. Rev. Microbiol., 61, 215-236. Hengge-Aronis, R., 2002: Recent insights into the general stress response regulatory network in Escherichia coli. J. Mol. Microbiol. Biotechnol., 4, 341-346. Hoang, T. T., R. R. Karkhoff-Schweizer, A. J. Kutchma, and H. P. Schweizer, 1998: A broadhost-range Flp- FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene, 212, 77-86. Hoffman, L. R., D. A. D'Argenio, M. J. MacCoss, Z. Zhang, R. A. Jones, and S. I. Miller, 2005: Aminoglycoside antibiotics induce bacterial biofilm formation. Nature, 436, 1171-1175. Houghton, J. L., K. D. Green, W. Chen, and S. Garneau‐Tsodikova, 2010: The future of aminoglycosides: the end or renaissance? ChemBioChem, 11, 880-902. Imlay, J. A., S. M. Chin, and S. Linn, 1988: Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science, 240, 640-642. 89 Islam, S., S. Jalal, and B. Wretlind, 2004: Expression of the MexXY efflux pump in amikacin‐ resistant isolates of Pseudomonas aeruginosa. Clin. Microbiol. Infec., 10, 877-883. Islam, S., H. Oh, S. Jalal, F. Karpati, O. Ciofu, N. Hoiby, and B. Wretlind, 2009: Chromosomal mechanisms of aminoglycoside resistance in Pseudomonas aeruginosa isolates from cystic fibrosis patients., Clin. Microbiol. Infec., 15, 60-66. Jacoby, G. A., M. J. Blaser, P. Santanam, H. Hachler, F. H. Kayser, R. S. Hare, and G. H. Miller, 1990: Appearance of amikacin and tobramycin resistance due to 4'-aminoglycoside nucleotidyltransferase [ANT (4')-II] in Gram-negative pathogens. Antimicrob. Agents Chemother., 34, 2381-2386. Jana, S., J. Deb, 2006: Molecular understanding of aminoglycoside action and resistance. Appl. Microbiol. Biotechnol., 70, 140-150. Jeannot, K., M. L. Sobel, F. El Garch, K. Poole, and P. Plésiat, 2005: Induction of the MexXY efflux pump in Pseudomonas aeruginosa is dependent on drug-ribosome interaction. J. Bacteriol., 187, 5341-5346. Jorgensen, F., M. Bally, V. Chapon-Herve, G. Michel, A. Lazdunski, P. Williams, and G. S. Stewart, 1999: RpoS-dependent stress tolerance in Pseudomonas aeruginosa. Microbiology, 145 (Pt 4), 835-844. Keen, N., S. Tamaki, D. Kobayashi, and D. Trollinger, 1988: Improved broad-host-range plasmids for DNA cloning in Gram-negative bacteria. Gene, 70, 191-197. Keren, I., Y. Wu, J. Inocencio, L. R. Mulcahy, and K. Lewis, 2013: Killing by bactericidal antibiotics does not depend on reactive oxygen species. Science, 339, 1213-1216. Kettner, M., J. Kallová, M. Hletkova, and P. Milosovic, 1995: Incidence and mechanisms of aminoglycoside resistance in Pseudomonas aeruginosa serotype O11 isolates. Infection, 23, 380383. Kindrachuk, K. N., L. Fernández, M. Bains, and R. E. Hancock, 2011: Involvement of an ATP90 dependent protease, PA0779/AsrA, in inducing heat shock in response to tobramycin in Pseudomonas aeruginosa. Antimicrob. Agents Chemother., 55, 1874-1882. Klauck, E., M. Lingnau, and R. Hengge‐Aronis, 2001: Role of the response regulator RssB in σS recognition and initiation of σS proteolysis in Escherichia coli. Mol. Microbiol., 40, 1381-1390. Kohanski, M. A., D. J. Dwyer, B. Hayete, C. A. Lawrence, and J. J. Collins, 2007: A common mechanism of cellular death induced by bactericidal antibiotics. Cell, 130, 797-810. Kohanski, M. A., M. A. DePristo, and J. J. Collins, 2010: Sublethal antibiotic treatment leads to multidrug resistance via radical-induced mutagenesis. Mol. Cell, 37, 311-320. Kotra, L. P., J. Haddad, and S. Mobashery, 2000: Aminoglycosides: perspectives on mechanisms of action and resistance and strategies to counter resistance. Antimicrob. Agents Chemother., 44, 3249-3256. Krahn, T., C. Gilmour, J. Tilak, S. Fraud, N. Kerr, C. H. F. Lau, and K. Poole, 2012: Determinants of intrinsic aminoglycoside resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother., 56, 5591–5602. Krahn, T., 2012: Chromosomal Determinants of Aminoglycoside Resistance in Pseudomonas aeruginosa. M.S. thesis, Dept. Biomedical and Molecular Science, Queen's University, 132pp, [Available online at http:// qspace.library.queensu.ca/ bitstream/1974/7513/3/ Krahn_ Thomas_ L_201209_MSc.pdf.] Lacalle, R. A., J. A. Tercero, J. Vara, and A. Jimenez, 1993: Identification of the gene encoding an N-acetylpuromycin N-acetylhydrolase in the puromycin biosynthetic gene cluster from Streptomyces alboniger. J. Bacteriol., 175, 7474-7478. Lagrange‐Puget, M., I. Durieu, R. Ecochard, F. Abbas‐Chorfa, J. Drai, J. Steghens, Y. Pacheco, D. Vital‐Durand, and G. Bellon, 2004: Longitudinal study of oxidative status in 312 cystic fibrosis patients in stable state and during bronchial exacerbation. Pediatr. Pulmonol., 38, 43-49. Lambert, T., G. Gerbaud, and P. Courvalin, 1994: Characterization of transposon Tn1528, which 91 confers amikacin resistance by synthesis of aminoglycoside 3'-O-phosphotransferase type VI. Antimicrob.Agents Chemother., 38, 702-706. Lau, C. H. F., S. Fraud, M. Jones, S. N. Peterson, and K. Poole, 2012: Reduced expression of the rplU-rpmA ribosomal protein operon in mexXY-expressing panaminoglycoside-resistant mutants of Pseudomonas aeruginosa. Antimicrob. Agents Chemother., 56, 5171-5179. Lau, C. H., S. Fraud, M. Jones, S. N. Peterson, and K. Poole, 2013: Mutational activation of the AmgRS two-component system in aminoglycoside-resistant Pseudomonas aeruginosa. Antimicrob. Agents Chemother., 57, 2243-2251. Leclerc, D., P. Melançon, and L. Brakier-Gingras, 1991: The interaction between streptomycin and ribosomal RNA. Biochimie, 73, 1431-1438. Lewis, K., 2008: Multidrug tolerance of biofilms and persister cells. Curr. Top. Microbiol. Immunol., 322, 107-131 Lewis, K., 2010: Persister cells. Annu. Rev. Microbiol., 64, 357-372. Li, X., H. Nikaido, 2009: Efflux-mediated drug resistance in bacteria. Drugs, 69, 1555-1623. Lister, P. D., D. J. Wolter, and N. D. Hanson, 2009: Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin. Microbiol. Rev., 22, 582-610. Liu, Y., J. A. Imlay, 2013: Cell death from antibiotics without the involvement of reactive oxygen species. Science, 339, 1210-1213. Livermore, D. M., 2002: Multiple mechanisms of antimicrobial resistance in Pseudomonas aeruginosa: our worst nightmare? Clin. Infect. Dis., 34, 634-640. Llano-Sotelo, B., E. F. Azucena Jr, L. P. Kotra, S. Mobashery, and C. S. Chow, 2002: Aminoglycosides modified by resistance enzymes display diminished binding to the bacterial ribosomal aminoacyl-tRNA site. Chem. Biol., 9, 455-463. 92 Loh, B., C. Grant, and R. E. Hancock, 1984: Use of the fluorescent probe 1-Nphenylnaphthylamine to study the interactions of aminoglycoside antibiotics with the outer membrane of Pseudomonas aeruginosa. Antimicrob. Agents Chemother., 26, 546-551. Lopez, D., H. Vlamakis, and R. Kolter, 2010: Biofilms. Cold Spring Harb. Perspect. Biol., 2, a000398. Lyczak, J. B., C. L. Cannon, and G. B. Pier, 2002: Lung infections associated with cystic fibrosis. Clin. Microbiol. Rev., 15, 194-222. Macia, M. D., D. Blanquer, B. Togores, J. Sauleda, J. L. Perez, and A. Oliver, 2005: Hypermutation is a key factor in development of multiple antimicrobial resistance in Pseudomonas aeruginosa strains causing chronic lung infections. Antimicrob. Agents Chemother., 49, 3382-3386. MacLeod, D. L., L. E. Nelson, R. M. Shawar, B. B. Lin, L. G. Lockwood, J. E. Dirks, G. H. Miller, J. L. Burns, and R. L. Garber, 2000: Aminoglycoside resistance mechanisms for cystic fibrosis Pseudomonas aeruginosa isolates are unchanged by long term, intermittent, inhaled tobramycin treatment. J. Infect. Dis., 181, 1180-1184. Magnet, S., J. S. Blanchard, 2005: Molecular insights into aminoglycoside action and resistance. Chem. Rev., 105, 477-498. Mah, T., B. Pitts, B. Pellock, G. C. Walker, P. S. Stewart, and G. A. O'Toole, 2003: A genetic basis for Pseudomonas aeruginosa biofilm antibiotic resistance. Nature, 426, 306-310. Margolis, P., A. Driks, and R. Losick, 1991: Establishment of cell type by compartmentalized activation of a transcription factor. Science, 254, 562-565. Martin, N. L., T. J. Beveridge, 1986: Gentamicin interaction with Pseudomonas aeruginosa cell envelope. Antimicrob. Agents Chemother., 29, 1079-1087. Masuda, N., E. Sakagawa, S. Ohya, N. Gotoh, H. Tsujimoto, and T. Nishino, 2000: Contribution 93 of the MexX-MexY-OprM efflux system to intrinsic resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother., 44, 2242-2246. Masuda, N., S. Ohya, 1992: Cross-resistance to meropenem, cephems, and quinolones in Pseudomonas aeruginosa. Antimicrob. Agents Chemother., 36, 1847-1851. Matsuo, Y., S. Eda, N. Gotoh, E. Yoshihara, and T. Nakae, 2004: MexZ‐mediated regulation of mexXY multidrug efflux pump expression in Pseudomonas aeruginosa by binding on the mexZ‐ mexX intergenic DNA. FEMS Microbiol. Lett., 238, 23-28. McCoy, L. S., Y. Xie, and Y. Tor, 2011: Antibiotics that target protein synthesis. Wiley Interdiscip. Rev. RNA, 2, 209-232. Mesaros, N., P. Nordmann, P. Plesiat, M. Roussel‐Delvallez, J. Van Eldere, Y. Glupczynski, Y. Van Laethem, F. Jacobs, P. Lebecque, and A. Malfroot, 2007: Pseudomonas aeruginosa: resistance and therapeutic options at the turn of the new millennium. Clin. Microbiol. Infec., 13, 560-578. Miller, G., F. Sabatelli, R. Hare, Y. Glupczynski, P. Mackey, D. Shlaes, K. Shimizu, and K. Shaw, 1997: The most frequent aminoglycoside resistance mechanisms changes with time and geographic Area: A reflection of aminoglycoside usage patterns? Clin. Infect. Dis, 24, S46-S62. Miller, G., F. Sabatelli, L. Naples, R. Hare, and K. Shaw, 1995: The most frequently occurring aminoglycoside resistance mechanisms-combined results of surveys in 8 regions of the world. J. Chemoter., 7, 17-30. Min, K., C. M. Hilditch, B. Diederich, J. Errington, and M. D. Yudkin, 1993: σF, the first compartment-specific transcription factor of B. subtilis, is regulated by an anti-σ factor that is also a protein kinase. Cell, 74, 735-742. Mine, T., Y. Morita, A. Kataoka, T. Mizushima, and T. Tsuchiya, 1999: Expression in Escherichia coli of a new multidrug efflux pump, MexXY, from Pseudomonas aeruginosa. Antimicrob. Agents Chemother., 43, 415-417. 94 Moazed, D., H. F. Noller, 1987: Interaction of antibiotics with functional sites in 16S ribosomal RNA. Nature, 327, 389-394. Morimoto, R. I., M. P. Kline, D. N. Bimston, and J. J. Cotto, 1997: The heat-shock response: regulation and function of heat-shock proteins and molecular chaperones. Essays Biochem., 32, 17-29. Morita, Y., M. L. Sobel, and K. Poole, 2006: Antibiotic inducibility of the MexXY multidrug efflux system of Pseudomonas aeruginosa: involvement of the antibiotic-inducible PA5471 gene product. J. Bacteriol., 188, 1847-1855. Mulcahy, H., L. Charron-Mazenod, and S. Lewenza, 2008: Extracellular DNA chelates cations and induces antibiotic resistance in Pseudomonas aeruginosa biofilms. PLoS pathog., 4, e1000213. Mulcahy, L. R., J. L. Burns, S. Lory, and K. Lewis, 2010: Emergence of Pseudomonas aeruginosa strains producing high levels of persister cells in patients with cystic fibrosis. J. Bacteriol., 192, 6191-6199. Nichols, W. W., S. N. Young, 1985: Respiration-dependent uptake of dihydrostreptomycin by Escherichia coli. Its irreversible nature and lack of evidence for a uniport process. Biochem. J., 228, 505-512. Nichols, W. W., S. M. Dorrington, M. P. Slack, and H. L. Walmsley, 1988: Inhibition of tobramycin diffusion by binding to alginate. Antimicrob. Agents Chemother., 32, 518-523. Ogle, J. M., A. P. Carter, and V. Ramakrishnan, 2003: Insights into the decoding mechanism from recent ribosome structures. Trends Biochem. Sci., 28, 259-266. Ogle, J. M., F. V. Murphy IV, M. J. Tarry, and V. Ramakrishnan, 2002: Selection of tRNA by the ribosome requires a transition from an open to a closed form. Cell, 111, 721-732. Oka, A., H. Sugisaki, and M. Takanami, 1981: Nucleotide sequence of the kanamycin resistance transposon Tn903. J. Mol. Biol., 147, 217-226. 95 Oliver, A., A. Mena, 2010: Bacterial hypermutation in cystic fibrosis, not only for antibiotic resistance. Clin. Microbiol. Infec., 16, 798-808. Oliver, A., F. Baquero, and J. Blázquez, 2002: The mismatch repair system (mutS, mutL and uvrD genes) in Pseudomonas aeruginosa: molecular characterization of naturally occurring mutants. Mol. Microbiol., 43, 1641-1650. Osmon, S., S. Ward, V. J. Fraser, and M. H. Kollef, 2004: Hospital mortality for patients with bacteremia due to Staphylococcus aureus or Pseudomonas aeruginosa. Chest, 125, 607-616. Pamp, S. J., M. Gjermansen, H. K. Johansen, and T. Tolker‐Nielsen, 2008: Tolerance to the antimicrobial peptide colistin in Pseudomonas aeruginosa biofilms is linked to metabolically active cells, and depends on the pmr and mexAB-OprM genes. Mol. Microbiol., 68, 223-240. Partridge, S., D. Foulger, and J. Errington, 1991: The role of σF in prespore‐specific transcription in Bacillus subtilis. Mol. Microbiol., 5, 757-767. Pednekar, S., V. Dohe, S. Deshpande, S. Kamble, R. Bharadwaj, and Y. Shouche, 2010: An outbreak of Pseudomonas aeruginosa in a burn unit. Burns, 36, e130-e131. Phillips, I., A. King, and K. Shannon, 1986: Prevalence and mechanisms of aminoglycoside resistance: a ten-year study. Am. J. Med., 80, 48-55. Poirel, L., T. Naas, D. Nicolas, L. Collet, S. Bellais, J. D. Cavallo, and P. Nordmann, 2000: Characterization of VIM-2, a carbapenem-hydrolyzing metallo-β-lactamase and its plasmid-and integron-borne gene from a Pseudomonas aeruginosa clinical isolate in France. Antimicrob. Agents Chemother., 44, 891-897. Poole, K., 2005a: Aminoglycoside resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother., 49, 479-487. Poole, K., 2005b: Efflux-mediated antimicrobial resistance. J. Antimicrob. Chemother., 56, 2051. 96 Poole, K., 2004: Efflux pumps. Pseudomonas. J. L. Ramos Ed., Springer, 635–674. Poole, K., 2007: Efflux pumps as antimicrobial resistance mechanisms. Ann. Med., 39, 162-176. Poole, K., 2011: Pseudomonas aeruginosa: resistance to the max. Front. Microbiol., 2, 65. Poole, K., 2012: Stress responses as determinants of antimicrobial resistance in Gram-negative bacteria. Trends in microbiology, 20, 227-234. Price, K., P. Kresel, L. Farchione, S. Siskin, and S. Karpow, 1981: Epidemiological studies of aminoglycoside resistance in the USA. J. Antimicrob. Chemother., 8, 89-105. Ramirez, M. S., M. E. Tolmasky, 2010: Aminoglycoside modifying enzymes. Drug. Resist. Update., 13, 151-171. Reyonolds, A. V., J. M. Hamilton-Miller, and W. Brumfitt, 1976: In vitro activity of amikacin and ten other aminoglycoside antibiotics against gentamicin-resistant bacterial strains. J. Infect. Dis., 134 (Suppl.), S291-S296. Rocke, D.M. and Durbin, B., 2001: A model for measurement error for gene expression arrays. J. Comp. Biol., 8, 557-569 Rodríguez Esparragón, F., M. González Martín, Z. González Lama, F. J. Sabatelli, and M. T. Tejedor Junco, 2000: Aminoglycoside resistance mechanisms in clinical isolates of Pseudomonas aeruginosa from the Canary Islands. Int. J. Med. Microbiol., 289, 817-826. Saavedra, S., D. Vera, and C. H. Ramírez-Ronda, 1986: Susceptibility of aerobic Gram-negative Bacilli to Aminoglycosides: Effects of 45 months of amikacin as first-linaminoglycoside therapy. Am. J. Med., 80, 65-70. Sabtcheva, S., M. Galimand, G. Gerbaud, P. Courvalin, and T. Lambert, 2003: Aminoglycoside resistance gene ant(4')-IIb of Pseudomonas aeruginosa BM4492, a clinical isolate from Bulgaria. Antimicrob. Agents Chemother., 47, 1584-1588. 97 Sambrook, J., D. W. Russell, and D. W. Russell, 2001: Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N. Y. Savli, H., A. Karadenizli, F. Kolayli, S. Gundes, U. Ozbek, and H. Vahaboglu, 2003: Expression stability of six housekeeping genes: A proposal for resistance gene quantification studies of Pseudomonas aeruginosa by real-time quantitative RT-PCR. J. Med. Microbiol., 52, 403-408. Schlacher, K., P. Pham, M. M. Cox, and M. F. Goodman, 2006: Roles of DNA polymerase V and RecA protein in SOS damage-induced mutation. Chem. Rev., 106, 406-419. Schmidt, R., P. Margolis, L. Duncan, R. Coppolecchia, C. P. Moran Jr, and R. Losick, 1990: Control of developmental transcription factor sigma F by sporulation regulatory proteins SpoIIAA and SpoIIAB in Bacillus subtilis. Proc. Natl. Acad. Sci. U.S.A., 87, 9221-9225. Schurek, K. N., A. K. Marr, P. K. Taylor, I. Wiegand, L. Semenec, B. K. Khaira, and R. E. W. Hancock, 2008: Novel genetic determinants of low-level aminoglycoside resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother., 52, 4213-4219. Schuster, M., A. C. Hawkins, C. S. Harwood, and E. Greenberg, 2004: The Pseudomonas aeruginosa RpoS regulon and its relationship to quorum sensing. Mol. Microbiol., 51, 973-985. Schuster, M. G., A. H. Norris, 1994: Community-acquired Pseudomonas aeruginosa pneumonia in patients with HIV infection. AIDS, 8, 1437-1442. Schweizer, H., 1991: Escherichia-Pseudomonas shuttle vectors derived from pUC18/19. Gene, 97, 109-112. Shakil, S., R. Khan, R. Zarrilli, and A. U. Khan, 2008: Aminoglycosides versus bacteria–a description of the action, resistance mechanism, and nosocomial battleground. J. Biomed. Sci., 15, 5-14. Shaw, K., P. Rather, R. Hare, and G. Miller, 1993a: Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol. Rev., 57, 138-163. 98 Shaw, K. J., R. S. Hare, F. J. Sabatelli, M. Rizzo, C. A. Cramer, L. Naples, S. Kocsi, H. Munayyer, P. Mann, and G. H. Miller, 1991: Correlation between aminoglycoside resistance profiles and DNA hybridization of clinical isolates. Antimicrob. Agents Chemother., 35, 22532261. Shaw, K. J., H. Munayyer, P. N. Rather, R. S. Hare, and G. H. Miller, 1993b: Nucleotide sequence analysis and DNA hybridization studies of the ant (4')-IIa gene from Pseudomonas aeruginosa. Antimicrob. Agents Chemother., 37, 708-714. Shaw, K. J., P. N. Rather, F. J. Sabatelli, P. Mann, H. Munayyer, R. Mierzwa, G. L. Petrikkos, R. S. Hare, G. H. Miller, and P. Bennett, 1992: Characterization of the chromosomal aac (6')-Ic gene from Serratia marcescens. Antimicrob. Agents Chemother., 36, 1447-1455. Shawar, R. M., D. L. MacLeod, R. L. Garber, J. L. Burns, J. R. Stapp, C. R. Clausen, and S. Tanaka, 1999: Activities of tobramycin and six other antibiotics against Pseudomonas aeruginosa isolates from patients with cystic fibrosis. Antimicrob. Agents Chemother., 43, 28772880. Silby, M. W., C. Winstanley, S. A. Godfrey, S. B. Levy, and R. W. Jackson, 2011: Pseudomonas genomes: diverse and adaptable. FEMS Microbiol. Rev., 35, 652-680. Silva, J. G., I. Carvalho, 2007: New insights into aminoglycoside antibiotics and derivatives. Curr. Med. Chem., 14, 1101-1119. Simon, R., U. Priefer, and A. Pühler, 1983: A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram-negative bacteria. Nat. Biotechnol., 1, 784791. Smith, E. E., D. G. Buckley, Z. Wu, C. Saenphimmachak, L. R. Hoffman, D. A. D'Argenio, S. I. Miller, B. W. Ramsey, D. P. Speert, S. M. Moskowitz, J. L. Burns, R. Kaul, and M. V. Olson, 2006: Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc. Natl. Acad. Sci. U.S.A., 103, 8487-8492. 99 Sobel, M. L., G. A. McKay, and K. Poole, 2003: Contribution of the MexXY multidrug transporter to aminoglycoside resistance in Pseudomonas aeruginosa clinical isolates. Antimicrob. Agents Chemother., 47, 3202-3207. Stanier, R. Y., N. J. Palleroni, and M. Doudoroff, 1966: The aerobic Pseudomonas: a taxonomic study. J. Gen. Microbiol., 43, 159-271. Strateva, T., D. Yordanov, 2009: Pseudomonas aeruginosa - a phenomenon of bacterial resistance. J. Med. Microbiol., 58, 1133-1148. Suh, S. J., L. Silo-Suh, D. E. Woods, D. J. Hassett, S. E. West, and D. E. Ohman, 1999: Effect of rpoS mutation on the stress response and expression of virulence factors in Pseudomonas aeruginosa. J. Bacteriol., 181, 3890-3897. Taber, H. W., J. Mueller, P. Miller, and A. Arrow, 1987: Bacterial uptake of aminoglycoside antibiotics. Microbiol. Rev., 51, 439-457. Taliaferro, D. L., P. J. Farabaugh, 2007: Testing constraints on rRNA bases that make nonsequence-specific contacts with the codon-anticodon complex in the ribosomal A-site. RNA, 13, 1279-1286. Tenover, F. C., K. L. Phillips, T. Gilbert, P. Lockhart, P. J. O'Hara, and J. J. Plorde, 1989: Development of a DNA probe from the deoxyribonucleotide sequence of a 3-N-aminoglycoside acetyltransferase [AAC (3)-I] resistance gene. Antimicrob. Agents Chemother., 33, 551-559. Tercero, J. A., J. C. Espinosa, R. A. Lacalle, and A. Jimenez, 1996: The biosynthetic pathway of the aminonucleoside antibiotic puromycin, as deduced from the molecular analysis of the pur cluster of Streptomyces alboniger. J. Biol. Chem., 271, 1579-1590. Thompson, R. C., D. B. Dix, R. B. Gerson, and A. M. Karim, 1981: Effect of Mg2+ concentration, polyamines, streptomycin, and mutations in ribosomal proteins on the accuracy of the two-step selection of aminoacyl tRNAs in protein biosynthesis. J. Biol. Chem., 256, 66766681. 100 Torres, C., M. H. Perlin, F. Baquero, D. L. Lerner, and S. A. Lerner, 2000: High-level amikacin resistance in Pseudomonas aeruginosa associated with a 3′-phosphotransferase with high affinity for amikacin. Int. J. Antimicrob Agents, 15, 257-263. Trunk, K., B. Benkert, N. Quäck, R. Münch, M. Scheer, J. Garbe, L. Jänsch, M. Trost, J. Wehland, and J. Buer, 2010: Anaerobic adaptation in Pseudomonas aeruginosa: definition of the Anr and Dnr regulons. Environ. Microbiol., 12, 1719-1733. Vakulenko, S. B., S. Mobashery, 2003: Versatility of aminoglycosides and prospects for their future. Clin. Microbiol. Rev., 16, 430-450. Vanhoof, R., E. Hannecart-Pokorni, and J. Content, 1998: Nomenclature of genes encoding aminoglycoside-modifying enzymes. Antimicrob. Agents Chemother., 42, 483. Vasil, M. L., 1986: Pseudomonas aeruginosa: Biology, mechanisms of virulence, epidemiology. J. Pediatr., 108, 800-805. Vijay, K., M. S. Brody, E. Fredlund, and C. W. Price, 2000: A PP2C phosphatase containing a PAS domain is required to convey signals of energy stress to the σB transcription factor of Bacillus subtilis. Mol. Microbiol., 35, 180-188. Vliegenthart, J., P. Ketelaar-van Gaalen, and J. van de Klundert, 1991: Nucleotide sequence of the aacC3 gene, a gentamicin resistance determinant encoding aminoglycoside-(3)-Nacetyltransferase III expressed in Pseudomonas aeruginosa but not in Escherichia coli. Antimicrob. Agents Chemother., 35, 892-897. Wagner, V. E., B. H. Iglewski, 2008: P. aeruginosa biofilms in CF infection. Clin. Rev. Allergy Immunol., 35, 124-134. Walsh, C., 2003: Antibiotics: actions, origins, resistance. American Society for Microbiology, 335 pp. 101 Wang, C., J. Jerng, K. Cheng, L. Lee, C. Yu, P. Hsueh, and P. Yang, 2006: Pandrug‐resistant Pseudomonas aeruginosa among hospitalised patients: clinical features, risk‐factors and outcomes. Clin. Microbiol. Infec., 12, 63-68. Wang, X., X. Zhao, M. Malik, and K. Drlica, 2010: Contribution of reactive oxygen species to pathways of quinolone-mediated bacterial cell death. J. Antimicrob. Chemother., 65, 520-524. Weng, L., Dai, H., Zhan Y., He, Y., Stepaniants, S. and Bassett, D. (2006) Rosetta error model for gene expression analysis. Bioinformatics, 22, 1111-1121. Werner, E., F. Roe, A. Bugnicourt, M. J. Franklin, A. Heydorn, S. Molin, B. Pitts, and P. S. Stewart, 2004: Stratified growth in Pseudomonas aeruginosa biofilms. Appl. Environ. Microbiol., 70, 6188-6196. Westbrock-Wadman, S., D. R. Sherman, M. J. Hickey, S. N. Coulter, Y. Q. Zhu, P. Warrener, L. Y. Nguyen, R. M. Shawar, K. R. Folger, and C. K. Stover, 1999: Characterization of a Pseudomonas aeruginosa efflux pump contributing to aminoglycoside impermeability. Antimicrob. Agents Chemother., 43, 2975-2983. Whiteley, M., M. R. Parsek, and E. Greenberg, 2000: Regulation of quorum sensing by RpoS in Pseudomonas aeruginosa. J. Bacteriol., 182, 4356-4360. Wilson, D. N., G. Blaha, S. R. Connell, P. V. Ivanov, H. Jenke, U. Stelzl, Y. Teraoka, and K. H. Nierhaus, 2002: Protein synthesis at atomic resolution: mechanistics of translation in the light of highly resolved structures for the ribosome. Curr. Protein Pept. Sci., 3, 1-53. Wohlleben, W., W. Arnold, L. Bissonnette, A. Pelletier, A. Tanguay, P. H. Roy, G. C. Gamboa, G. F. Barry, E. Aubert, and J. Davies, 1989: On the evolution of Tn21-like multiresistance transposons: sequence analysis of the gene (aacC1) for gentamicin acetyltransferase-3-I (AAC (3)-I), another member of the Tn21-based expression cassette. Mol. gen. genet., 217, 202-208. 102 Wood, L. G., D. A. Fitzgerald, P. G. Gibson, D. M. Cooper, C. E. Collins, and M. L. Garg, 2001: Oxidative stress in cystic fibrosis: dietary and metabolic factors. J. Am. Coll. Nutr., 20, 157-165. Wright, G. D., 1999: Aminoglycoside-modifying enzymes. Curr. Opin. Microbiol., 2, 499-503. Yamamoto, M., A. Ueda, M. Kudo, Y. Matsuo, J. Fukushima, T. Nakae, T. Kaneko, and Y. Ishigatsubo, 2009: Role of MexZ and PA5471 in transcriptional regulation of mexXY in Pseudomonas aeruginosa. Microbiology, 155, 3312-3321. Yang, X., C. M. Kang, M. S. Brody, and C. W. Price, 1996: Opposing pairs of serine protein kinases and phosphatases transmit signals of environmental stress to activate a bacterial transcription factor. Genes Dev., 10, 2265-2275. Yokoyama, K., Y. Doi, K. Yamane, H. Kurokawa, N. Shibata, K. Shibayama, T. Yagi, H. Kato, and Y. Arakawa, 2003: Acquisition of 16S rRNA methylase gene in Pseudomonas aeruginosa. Lancet, 362, 1888-1893. Yoshimura, F., H. Nikaido, 1982: Permeability of Pseudomonas aeruginosa outer membrane to hydrophilic solutes. J. Bacteriol., 152, 636-642. Young, S. A., F. C. Tenover, T. D. Gootz, K. P. Gordon, and J. J. Plorde, 1985: Development of two DNA probes for differentiating the structural genes of subclasses I and II of the aminoglycoside-modifying enzyme 3'-aminoglycoside phosphotransferase. Antimicrob. Agents Chemother., 27, 739-744. Zhou, Y., H. Yu, Q. Guo, X. Xu, X. Ye, S. Wu, Y. Guo, and M. Wang, 2010: Distribution of 16S rRNA methylases among different species of Gram-negative bacilli with high-level resistance to aminoglycosides. Eur. J. Clin. Microbiol., 29, 1349-1353. Zumft, W. G., 1997: Cell biology and molecular basis of denitrification. Microbiol. Mol. Biol. Rev., 61, 533-616. 103