Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Psychoneuroimmunology wikipedia , lookup
Rheumatic fever wikipedia , lookup
Polyclonal B cell response wikipedia , lookup
Kawasaki disease wikipedia , lookup
Monoclonal antibody wikipedia , lookup
Childhood immunizations in the United States wikipedia , lookup
Behçet's disease wikipedia , lookup
Anti-nuclear antibody wikipedia , lookup
Molecular mimicry wikipedia , lookup
Globalization and disease wikipedia , lookup
Hygiene hypothesis wikipedia , lookup
Germ theory of disease wikipedia , lookup
Schistosomiasis wikipedia , lookup
African trypanosomiasis wikipedia , lookup
Management of multiple sclerosis wikipedia , lookup
Neuromyelitis optica wikipedia , lookup
Rheumatoid arthritis wikipedia , lookup
Autoimmune encephalitis wikipedia , lookup
Multiple sclerosis research wikipedia , lookup
Immunosuppressive drug wikipedia , lookup
Hepatitis B wikipedia , lookup
Autoimmunity wikipedia , lookup
Sjögren syndrome wikipedia , lookup
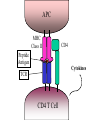
Biliary obstruction, autoimmune diseases of the liver John J O’Leary Learning objectives • Understand bile duct obstruction [causes and effects] • Understand what is meant by the term auto-immune liver disease: – Primary biliary cirrhosis [PBC] – Primary sclerosing cholangitis [PSC] – Autoimmune hepatitis [AIH] Biliary obstruction Pathological effects of biliary obstruction Biliary obstruction High local concentration of bile salts Inflammation Fibrosis & scarring Biliary fistula Fibrosis and scarring Biliary stasis Repeated cholangitis Liver atrophy Biliary cirrhosis & portal HTN (very late) Causes of Benign strictures Causes of Malignant strictures Symptoms and history: Biliary colic Obstructive jaundice Cholangitis – Charcot’s triad jaundice; fever, usually with rigors; and right upper quadrant abdominal pain. – Reynold’s pentad + hypotension and an altered mental state Past history of cholecystectomy/ other GI surgery (eventful) History s/o pancreatitis, trauma, radiation, alcohol abuse Signs: Icterus s/o hepatocellular failure Courvoisier's sign e/o biliary fistula, peritonitis, biloma Nutritional deficiencies pale stools dark urine itchiness (pruritus) HISTOPATHOLOGY Under a microscope, the individual hepatocytes will have a brownish-green stippled appearance within the cytoplasm, representing bile that cannot get out of the cell. Canalicular bile plugs between individual hepatocytes or within bile ducts may also be seen, representing bile that has been excreted from the hepatocytes but cannot go any further due to the obstruction. When these plugs occur within the bile duct, sufficient pressure (caused by bile accumulation) can cause them to rupture, spilling bile into the surrounding tissue, causing hepatic necrosis. These areas are known as bile lakes, and are typically seen only with extra-hepatic obstruction. Strasberg classification of biliary injury & stricture Laboratory investigations Bilirubin Alkaline phosphatase & PT Anemia, amylase & lipase, GGT ESR, LDH Tumor markers: CA 19-9, CEA, AFP Imaging studies: Ultrasound CT scan MRCP HIDA scan ERCP Endoscopic ultrasound PTC Fistulography Ultrasonography: Detects intra/extra hepatic ductal dilatation Less accuracy in defining etiology Sensitivity – 94% if bilirubin > 10mg% – 47% otherwise Endoscopic ultrasound: • Extra hepatic bile duct readily visualized • Detects choledocholithiasis (>95%) • Sensitive in diagnosis & staging of malignancy CT Scan: • Highly sensitive (esp. with contrast) • Detects site & cause of obstruction • Better anatomical delineation • CT cholangiography MRI & MRCP: • Bile – high signal intensity on T2 weighted images • Visualises – biliary dilatation (97100%), site of obstruction (87%), hepatic parenchyma & vasculature • Only diagnostic HIDA Scan [Hepatobiliary Imino-Diacetic Acid scan] • Helps in diagnosing biliary leaks • Provides functional assessment of incomplete strictures and surgical anastomosis • suggest complete biliary obstruction if the small intestine is not visualized in 60 minutes • insensitive for helping detect biliary dilatation or the site and cause of bile duct obstruction Cholangiography: Gold standard Endoscopic/ percutaneous Features: – define the anatomy of the proximal biliary tree – decompression of the biliary system – Non-operative dilation of bile duct strictures – allow for the simultaneous placement of drainage catheters – of assistance with regard to surgical reconstruction Treatment: Options: Endoscopic or percutaneous balloon dilatation and insertion of an endoprosthesis Surgery Pre-procedure antibiotic prophylaxis RX of malignant strictures: Depends on: resectability & stage general condition Resectable (15-20%) – Radical resection with biliary enteric anastomosis Palliative – endoscopic stenting – percutaneous transhepatic stenting – Palliative resection with surgical bypass (mortality – 33%) RX of benign strictures: Operative management – to surgically re-establish bile flow within the biliary tree and into the proximal gastrointestinal tract in a manner that prevents cholestasis, cholangitis, sludge and stone formation, restricture, or biliary cirrhosis Non–operative management – to correct the increased resistance to biliary flow caused by a reduction in the diameter of the lumen Autoimmune diseases of the liver Autoimmune liver disease • Primary biliary cirrhosis • Primary sclerosing cholangitis • Autoimmune hepatitis Primary biliary cirrhosis PRIMARY BILIARY CIRRHOSIS Primary biliary cirrhosis is an autoimmune disease of the liver marked by: - the slow progressive destruction of the small bile ducts (bile canaliculi) within the liver. - when these ducts are damaged, bile builds up in the liver (cholestasis) and over time damages the tissue. - this can lead to scarring, fibrosis and cirrhosis. - it was previously thought to be a rare disease, but more recent studies have shown that it may affect up to 1 in 3-4,000 people; the sex ratio is at least 9:1 (women to men) In some areas of the US and UK the prevalence is estimated to be as high as 1 in 4000. This is much more common than in South America or Africa, which may be due to better recognition in the US and UK. First-degree relatives may have as much as a 500 times increase in prevalence, but there is debate if this risk is greater in the same generation relatives or the one that follows. After liver transplant, the recurrence rate may be as high as 18% at 5 years, and up to 30% at 10 years. There is no consensus on risk factors for recurrence of the disease The following signs may be present in PBC: • Fatigue • Pruritus (itchy skin) • Jaundice (yellowing of the eyes and skin) • Xanthelasmata (focal collections of cholesterol in the skin, especially around the eyes) • Complications of cirrhosis and portal hypertension: • Fluid retention in the abdomen (ascites) • Hypersplenism • Esophageal varices • Hepatic encephalopathy, up to coma, in extreme cases. • Association with extra-hepatic autoimmune disorder[s] such as Rheumatoid arthritis or Sjögren's syndrome (up to 80% incidence). To diagnose PBC, distinctions should be established from other conditions with similar symptoms, such as autoimmune hepatitis or primary sclerosing cholangitis (PSC). Diagnostic blood tests include: • Abnormal liver function tests (high alkaline phosphatase, elevated AST, ALT) • Presence of certain antibodies: antimitochondrial antibody, antinuclear antibody (the M2-IgG antimitochondrial antibody is the most specific test) • Abdominal ultrasound or a CT scan is usually performed to rule out blockage to the bile ducts. Previously most suspected sufferers underwent a liver biopsy, and - if uncertainty remained - endoscopic retrograde cholangiopancreatography (ERCP, an endoscopic investigation of the bile duct). Now most patients are diagnosed without invasive investigation since the combination of anti-mitochondrial antibodies (see below) and typical (cholestatic) liver function tests are considered diagnostic. However, a liver biopsy is necessary to determine the stage of disease. Anti-nuclear antibodies appear to be prognostic agents in PBC. Anti-glycoprotein-210 antibodies, and to a lessor degree anti-p62 antibodies correlate with progression toward end stage liver failure. Anti-centromere antibodies correlate with developing portal hypertension. Anti-np62 and anti-sp100 are also found in association with PBC. STAGES OF DISEASE • Stage 1 - Portal Stage: Normal sized triads; portal inflammation, subtle bile duct damage. Granulomas are often detected in this stage. • Stage 2 - Periportal Stage: Enlarged triads; periportal fibrosis and/or inflammation. Typically characterized by the finding of a proliferation of small bile ducts. • Stage 3 - Septal Stage: Active and/or passive fibrous septa. • Stage 4 - Biliary Cirrhosis: Nodules present; garland or jigsaw pattern. The cause of the disease is unknown at this time - an immunological basis for the disease, making it an autoimmune disorder. - most patients (>90%) have anti-mitochondrial antibodies (AMAs) against pyruvate dehydrogenase complex (PDC-E2), an enzyme complex that is found in mitochondria. - an increase in GGT could indicate presence of Primary Biliary Cirrhosis. - 57% of patients with acute liver failure have anti-transglutaminase antibodies suggesting a role of gluten sensitivity in primary biliary cirrhosis, and primary biliary cirrhosis is considerably more common in gluten sensitive enteropathy than the normal population. - in some cases of disease protein expression may cause an immune tolerance failure, as might be the case with gp210 and p62, nuclear pore proteins. Gp210 has increased expression in the bile duct of anti-gp210 positive patients. - Both proteins appear to be prognostic of liver failure relative to antimitochondrial antibodies. Course and cure of the disease: - no known cure, but medication may slow the progression - ursodeoxycholic acid (Ursodiol) is the most frequently used treatment. This helps reduce the cholestasis and improves blood test results (liver function tests). - to relieve itching caused by bile acids in circulation, which would normally be removed by the liver, cholestyramine (a bile acid sequestrant) may be prescribed to absorb bile acids in the gut and be eliminated, rather than re-enter the blood stream. - alcoholic beverages are contraindicated. - in advanced cases, a liver transplant, if successful, results in a favourable prognosis. - multivitamins (esp. Vitamin D) and calcium are also recommended as patients with PBC have poor lipiddependent absorption of Vitamins A, D, E, K. Contrast-enhanced helical CT image obtained through liver during hepatic arterial phase shows several small, homogeneously enhancing lesions (arrows). Multiple lesions were seen throughout remainder of liver as well. Primary sclerosing cholangitis Primary sclerosing cholangitis (PSC) is a chronic liver disease caused by progressive inflammation and scarring of the bile ducts of the liver. The inflammation impedes the flow of bile to the gut, which can ultimately lead to liver cirrhosis and liver failure. The underlying cause of the inflammation is believed to be autoimmunity. The definitive treatment is liver transplantation. The cause(s) for PSC are unknown. It is often considered to be an autoimmune disorder. PSC is associated with inflammatory bowel disease and particularly ulcerative colitis, which coexists in approximately 70% of patients. Conversely, the prevalence of PSC in ulcerative colitis patients is ~4%. There is a 2:1 male-to-female predilection of PSC. There is an increased prevalence of HLA alleles A1, B8, and DR3 in PSC. The disease normally starts from age 30 to 60, though may begin in childhood. PSC progresses slowly, so the disease can be active for a long time before it is noticed or diagnosed. Signs and symptoms • Fatigue • Severe jaundice with intense itching • Malabsorption (especially of fat) and steatorrhea, leading to decreased levels of the fat-soluble vitamins, A, D, E and K. • Signs of cirrhosis • Ascending cholangitis, or infection of the bile duct. • Pale stools due to biliary obstruction • Dark urine due to excess conjugated bilirubin, which is water soluble, being excreted by the kidneys Diagnosis: - imaging of the bile duct [ERCP], - magnetic resonance cholangio-pancreatography (MRCP), - approximately 80% of patients have perinuclear antineutrophil cytoplasmic antibodies, also called p-ANCA, however this finding is not specific for PSC. - antinuclear antibodies and anti-smooth muscle antibodies are found in 20%-50% of PSC patients and, likewise, are not specific for the disease. - full blood count, liver enzymes, bilirubin levels (usually grossly elevated), renal function, electrolytes. - faecal fat determination is occasionally ordered when the symptoms of malabsorption are prominent. The differential diagnosis can include: primary biliary cirrhosis, drug induced cholestasis, cholangiocarcinoma, and HIV-associated cholangiopathy. Treatment: - ursodiol, a bile acid naturally produced by the liver, which has been shown to lower elevated liver enzyme numbers in people with PSC, but has not yet been proven effective at prolonging the life of the liver. - medication to relieve itching (antipruritics) and bile acid sequestrants (cholestyramine), antibiotics to treat infections, and vitamin supplements, as people with PSC are often deficient in vitamin A, vitamin D, vitamin E and vitamin K. - in some cases, ERCP, which may involve stenting of the common bile duct, may be necessary in order to open major blockages (dominant strictures). - Liver transplantation is the only proven long-term treatment of PSC. Indications for transplantation include recurrent bacterial cholangitis, jaundice refractory to medical and endoscopic treatment, decompensated cirrhosis and complications of portal hypertension. PSC is associated with cholangiocarcinoma Primary sclerosing cholangitis. Single-shot fast spin-echo MRCP image shows multifocal strictures and dilatations of the intrahepatic bile ducts Autoimmune hepatitis Overview • • • • • • • • Definition Prevalence Clinical manifestations Pathogenesis Subtypes Diagnosis Prognostic indices Treatment Autoimmune Hepatitis • self-perpetuating hepatocellular inflammation • unknown cause • characterization: – histologic features of interface hepatitis – hypergammaglobulinemia – serum autoantibodies • affects all ages, may be asymptomatic, frequently has an acute onset, and can also present as fulminant hepatitis. • Autoimmune hepatitis has an incidence of 1-2 per 100,000 per year, and a prevalence of 10-20/100,000. As with most other autoimmune diseases, it affects women much more often than men (70%). Prevalence • Mean annual incidence among white northern Europeans: 1.9 cases per 100,000 per year • US: 100,000-200,000 people • W>M 3.6:1, unexplained. • all ages, ethnic groups. • Frequency of AIH among pts with chronic liver disease in North America is 11-23% • Accounts for 5.9% in the National Institutes of Health Liver transplantation database. • Prevalence of AIH is greatest among northern European white groups who have a high frequency of HLA-DR3 and HLA-DR4 Clinical manifestations Pathogenesis • Unknown mechanism • Most popular hypotheses:interactive factors: – – – – – Triggering agent A genetic predisposition various determinants of autoantigen display immunocyte activation effector cell expansion. Pathogenesis • Triggering agents – infectious agents, drugs, toxins. – Multiple agents suggest that triggering epitope is a short amino acid sequence that is common in many antigens. – Possible long lag time b/w exposure to the trigger and onset of the disease – Triggering factor may not be needed for perpetuation of the disorder. Pathogenesis • Loss of self-tolerance – – molecular mimicry of a foreign antigen – self antigen • Only the cytochrome monooxygenase P-450 IID6 (CYP2D6) has been recognized as an autoantigen. – multiple self-antigens or foreign antigens may satisfy the minimal structural requirements and serve as immunogenic peptides Pathogenesis • • genetic predisposition Susceptibility HLA allelles encoding MHC class II: – influences the presentation of these autoantigens to CD4+ helper T cell thereby initiating an immune response. – the initiation of the immune response is dependent on the antigen binding groove of the class II MHC molecule. The sequence of amino acids that make up this antigen binding groove is encoded by a person’s HLA alleles. Thus, there are specific alleles that make a person more susceptible to developing AIH by influencing the immune response, and in turn the clinical manifestations and behavior of AIH. • Autoimmune promoters: – polymorphisms for TNF-α gene and cytotoxic T lymphocyte antigen 4 gene have been associated with increased immune reactivity and disease severity of AIH type I – Other: Vitamin D receptor (VDR) gene, point mutation of the tyrosine phosphatase CD45 gene, polymorphisms of the Fas gene, MHC class 1 chain-related A gene. APC MHC Class II CD4 Peptide Antigen Cytokines TCR CD4 T Cell Pathogenesis • Mechanism of Liver cell destruction via cellular and humoral mechanisms – Cell-mediated cytotoxicity • TH1 response (IL-2, IFN-γ, TNF-α) clonal expansion of cytotoxic T lymphocytes that infiltrate and destroy hepatocytes. • Genetic polymorphism that affects TNF- production may facilitate this pathway. – Antibody-dependent cell-mediated cytotoxicity • TH2 response (IL-4,5,6,8,10,13) B cell stimulation Ab production immunocyte complexes on hepatocyte surface targeted by NKT cells • Anti-inflammatory effects that counters TH1 action – Combination of mechanisms • Predominant mechanism depends on the phenotypic differentiation of CD4+ helper T cell, which in turn reflects the cytokine milieu, which in itself reflects the polymorphisms of the cytokine genes that favor excessive production of some modulators, such as TNF-, or deficient in others. AIH subtypes • • • • • AIH type 1 AIH type 2 AIH type 3 on the basis of immunoserologic markers. The International Autoimmune Hepatitis Group has not endorsed this subclassification. • Used mainly for descriptive value AIH type 1 • Most common form worldwide • characterized by the presence or absence of SMA and/or ANA in serum • Surrogate markers: Peri-nuclear anti-neutrophil cytoplasmic antibodies which occur in PSC and chronic UC, are found in 90% of patients who have type 1 AIH. • Bimodal age distribution (10-20; 45-70) • Female:male 3.6:1 • Risk factors for type 1 AIH – in whites of northern European descent [HLA-DR3 (DRB1*0301)] and –DR4 (DRB1*0401)] AIH type 1 • Associated with concurrent extra-hepatic diseases – Autoimmune thyroiditis (12%) – Graves disease (6%) – Chronic UC (6%) * (cholangiography to exclude PSC) – <1% RA, pernicious anemia, systemic sclerosis, Coomb’s test-positive HA, ITP, symptomatic cryoglobulinemia, leukocytoclastic vasculitis, nephritis, erythema nodosum, SLE, fibrosing alveolitis. • 40% of AIH type 1 presents with an acute onset of symptoms/signs indistinguishable from that of acute viral or toxic hepatitis and the disease may appear fulminant in fashion. • target autoantigen is unknown, but ASGPR (asialoglycoprotein receptor) found on hepatocyte surface is a candidate • Responds well to glucocorticoids AIH type 2 • • • • • • • • Characterized by presence of anti-LKM1 (liver/kidney microsome type 1) in serum P-ANCA is not found Mainly children (2-14 yo) but also seen in adults (in Europe, 20% of pts are adults; in US, 4% of pts are >18 yrs) Only AIH with an identified target autoantigen: cytochrome monooxygenase P-450 IID6 (CYP2D6) found in the cytosol of hepatocytes. – Recognized homologies b/w epitopes of CYP2D6 and genome of HCV. – <10% of Europeans with chronic Hep C have detectable antiLKM1 – Suggests molecular mimicry and antibody cross-reactivity, multiple exposures to virus mimicking self may be a way to break self-tolerance and induce AIH type 2. – Anti-LKM1 + pts with chronic Hep C in US pts is rare – differences in indigenous virus or host susceptiblity? Acute or fulminant presentation is possible – Thus essential to screen all pts who have an acute decompensation for type-specific autoantibodies. Associated with HLQA-B14, -DR3, -C4A-QD Susceptibility factor in German and Brazilians: DRB1*07 Like AIH type 1, also responds well to glucocorticoids AIH type 2 • • • • • • • • Distinct form of anti-LKM positive AIH Occurs in association with autoimmune polyendocrinopathy disorder (APECED) aka Polyglandular autoimmune syndrome type I (APS1) rare autosomal recessive disorder Caused by a signal gene mutation of the APS1 gene which encodes a transcription factor, autoimmune regulator (AIRE) which is expressed in epithelial and dendritic cells within the thymus where it regulates clonal deletion of autoreactive T cells, thus can affect self tolerance Features of this disease are ectodermal dystrophy, mucocutaneous candidiasis, multiple endocrine gland failure (parathyroids, adrenals, ovaries) Marked by the presence of numerous organ and non-organ specific autoantibodies and multiple concurrent autoimmune diseases. most common among Finns, Sardinians, and Iranian Jews Pts with APECED and AIH have an aggressive liver disease that does not respond well to standard immunosuppressive regimens. AIH type 3 • Least established form of the disease • Designation largely abandoned • Characterized by presence of antibodies to soluble liver antigen and liver/pancreas (anti-SLA, anti- LP) • 30-50 yo • Target autoantigens: thought to be Glutathione Stransferase, but a transfer ribonucleoprotein (tRNP) 50-kd protein was described in 2000 as the more likely target. • Clinical and laboratory features that are indistinguishable from AIH type 1 • Also responds well to glucocorticoids AIH subtypes Variant forms Diagnosis • Determination of serum aminotransferase and –globulin levels • Rule out ddx • Detection of ANA and /or SMA, or in their absence, anti-LKM1 • Liver tissue examination Diagnosis • determination of serum aminotransferase and –globulin levels – Predominant serum aminotransferase abnormality – Hypergammaglobulinemia • exclusion of other chronic liver diseases that have similar features – hereditary causes: (Wilson disease, alpha1-antitrypsin deficiency, genetic hemochromatosis – infectious causes: chronic viral hepatitis A, B, C – drug-induced liver disease (ETOH, minocycline, nitrofurantoin, INH, propylthiouracil, methyldopa) – NASH – immune cholangiopathies of PBC, PSC, autoimmune cholangitis Diagnosis • Detection of ANA and /or SMA, or in their absence, anti-LKM1 – conventional immunoserologic tests for AIH: • antinuclear antibodies (ANA), – present alone (13%), along with SMA (54%) – also be found in PBC, PSC, chronic viral hepatitis, drug-related hepatitis, NASH, alcohol-induced liver disease • smooth muscle antibodies (SMA), – Directed against actin and nonactin components (tubulin, vimentin, desmin, skeletin) – present (87%), sole marker (33%), with ANA (54%) • antibodies to liver/kidney microsome type 1 (anti-LKM1) – Typically occurs in absence of SMA and ANA – rare in the US (4% of adults with AIH in US) • perinuclear anti-neutrophil cytoplasmic antibodies (pANCAs) are common in type 1 AIH, – useful in evaluation patients who lack conventional autoantibodies. – Used to reclassify patients with cryptogenic chronic hepatitis as AIH, but they have not been formally assimilated into the diagnostic algorithm – Do not have diagnostic specificity nor do they have prognostic implications. Diagnosis • Detection of ANA and /or SMA, or in their absence, anti-LKM1 – NEW antibodies: • investigational in nature, but sufficient promise to support a probable diagnosis, not generally available, assays are not standardized • actin (anti-actin) – has less sensitivity, but greater specificity than SMA for AIH type 1 • asialoglycoprotein receptor (anti-ASGPR) – transmembrane glycoprotein on the hepatocyte surface which can capture, internalize, display potential antigens – Seen in AIH type 1 • soluble liver antigen/liver-pancreas (A) – Seen in AIH type 3 – used to reclassification of patients with cryptogenic chronic hepatitis as AIH • liver cytosol type 1 (anti-LC1) – Have been proposed as an antigenic target. – Seen in AIH type 2 – Prevalence is higher in pts < 20, Rare in pts >40 Diagnosis • Liver tissue examination – Liver bx is essential to establish diagnosis and assess disease severity to determine need for treatment – histological features of interface hepatitis (hallmark of the syndrome) – portal plasma cell infiltration typifies the disorder • lack of portal plasma cell infiltration does not preclude dx – 1999 update: lobular hepatitis is now part of histologic spectrum – aminotransferase and gamma-globulin levels do not predict histologic pattern of injury or the presence or absence of cirrhosis. – Histologic changes, such as ductopenia or destructive cholangitis, may indicate a variant syndrome of AIH and PSC, AIH an PBC, or autoimmune cholangitis. – Findings of steatosis or iron overload may suggest alternative diagnoses: • NASH • Wilson disease • Chronic Hep C • Drug toxicity • genetic hemochromatosis Interface hepatitis Here you can see the limiting plate of the portal tract, demarcating the hepatocyte boundary, is disrupted by a lymphoplasmacytic infiltrate. This histological pattern is the hallmark of autoimmune hepatitis, but it is not disease specific. Plasma cell infiltration Lobular Hepatitis PBC Marked mononuclear cell infiltrate surrounding and destroying a bile duct in primary biliary cirrhosis PSC Mononuclear cell infiltration and characteristic concentric fibrosis around a small bile duct in PSC Diagnostic Criteria Scoring System Prognosis Prognosis • 40% develop with cirrhosis – 54% develop esophageal varices w/in 2 years – 20% die from variceal hemorrhage if they don’t receive any treatment – hepatocellular carcinoma can occur in this pts but risk is small. • • • Presence of ascites or hepatic encephalopathy identifies pts with a poor prognosis. 13-20% of patients can have spontaneous resolution regardless of disease activity. A critical determinant of survival in the untreated patient is early tolerance of the disease. – Of pts who survive the early, most active stage of the disease, inactive cirrhosis develops in 41% of pts. – Untreated patients who have initial severe disease and survive the first 2 years of illness typically survive long term Treatment Indication for liver transplantation • Pts’ with ascites and hepatic encephalopathy identifies pt’s with poor prognosis. They can still respond to glucocorticoid therapy and should be treated before a decision regarding liver transplantation is made. • Indicated in decompensated pt with hepatic encephalopathy, ascites, and/or variceal hemorrhage during therapy for treatment failure. • Effective in pts who deteriorate during or after corticosteroid tx. • After transplantation, the autoantibodies and hypergammaglobulinemia disappear within 2 years the 5 year survival rate is 96%. Actuarial 10 year survival 75%. • Recurrent disease after transplantation is common but has been described mainly in patients who have inadequate immunosuppression. • RARE cases (3-5%) pts can develop AIH de novo after undergoing transplantation for non-autoimmune liver disease. Immunosuppression with CyA is a common feature. TX with prednisone or azathioprine is effective. AIH Summary • • • • • • • self-perpetuating hepatocellular inflammation of unknown cause Genetic predispostion Diverse clinical features 3 subtypes AIH and variants Dx: – interface hepatitis – hypergammaglobulinemia – serum autoantibodies – Rule out other liver diseases Consider in all pts with acute & chronic liver diseases and those with allograft dysfunction after transplantation Prednisone, Azathioprine • Evolving treatment options