Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Meeting on Cenibacterium arsenoxidans annotation - 14/04/05
Bacterial genome annotation in
the AGC group
Claudine Médigue
Atelier de Génomique Comparative
GENOSCOPE/CNRS UMR “Structure et évolution des génomes”
Dir. Jean Weissenbach
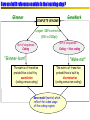
What is genome annotation ?
Annotation: A note, added by way of comment, or explanation.
Typical genome annotation questions:
What genes does this genome contain?
What is their location?
What proteins do they encode?
How are they regulated?
In what interactions and in what pathways do the
protein products participate?
What is genome annotation ?
Three annotation level
Syntaxic/structural annotation
• Location of genes (both proteincoding genes and RNA genes)
• Location of regulatory signals
• Location of other regions (such
as repeats, etc)
EMBL
Static view of the
genome
détection par contenu
Functionnal annotation
• Biological function of the genes
SWISSPROT
• Operators family
Process annotation (or relationnal)
Dynamic view
of the genome
How genomic objets are linked to build functionnal
module, responsible for specific task in the cell
such as :
• metabolic networks
• regulatory processes
• molecular assembly
•…
Experimental
results
L. Stein (2001)
Structural annotation tools
Oriloc : Cumulatif GC skew to predict the replication origin and terminus
tRNA-scan : tRNA gene prediction (G. Fichant et al.)
findrRNA
: rRNA gene finding
AMIGene : CDS prediction in bacterial genomes
ProFED
: Procaryotic
Frameshift Error Detection
AFC/Kmean : Statistical analysis (i.e, codon or oligonucleotide usage)
AMIMat : CDS prediction in bacterial genomes
Petrin : rho indépendant terminators prediction (C. Term et al.)
Spat : Pattern finding such as RBS, promoters, …(A. Viari et al.)
Nosferatu : Closest or distants DNA repeats (E. Rocha et al.)
From different authors
From the AGC group
Gene finding process
ORF (Open Reading Frame)
GTGGAATTGTGAGCGGATAACAATTTCACACAGGAAACAGCTATGACCGTGATTTTGGATTCA...GTCGTTTAACAACGTCG
Stop A D N N S T Q E T A M T V I T D S
V V Stop
=>ORF more than 300 nt in length: probably not a random ORF
Potential coding region
GTGGAATTGTGAGCGGATAACAATTTCACACAGGAAACAGCTATGACCGTGATTTTGGATTCA...GTCGTTTAACAACGTCG
Stop
M T V I T D S
V V Stop
Coding probablility ?
GTGGAATTGTGAGCGGATAACAATTTCACACAGGAAACAGCTATGACCGTGATTTTGGATTCA...GTCGTTTAACAACGTCG
Stop
M T V I T D S
V V Stop
rbs
Start candidates
=> We used a statistical property of coding regions based on different
compositions in oligonucleotides of length k between coding/non
coding region.
Ribosome binding sites (RBS)
Start
codon
http://cwx.prenhall.com/horton/medialib/media_portfolio/
RBS-finder (TIGR)
Gene finding : methods based on Markov Models
• Statistical model
A,C,G,T
k
The probability that a nucleotide is in
position I depends only on the type of
the k preceeding nucleotides :
P(X/X1...Xk)
i
Transition probabilities
Learning step => Gene models
• Practical use
phase 1
start
stop
Pcodant
Searching for stop/start codon patterns
(RBS) + chaining constraints
+3
+2
+1
-1
-2
-3
phase 2
w
phase 3
GeneMark (Borodovski)
Glimmer (Salzberg)
How are built reference models in the learning step ?
Glimmer
GeneMark
COMPLETE GENOME
Longest ORFs extraction
(500 to 1000pb)
Set of sequences :
Coding
Set of sequences :
Coding + Non coding
"Glimmer-learn"
"Make-mat"
The matrix of transition
probabilities is built by
assimilation
(coding versus coding)
The matrix of transition
probabilities is built by
discrimination
(coding versus non coding)
Gene model (matrix) which
reflect the codon usage
of the coding regions
Example of gene prediction
E. coli
+3
+2
+1
E. coli gene model
C. jejuni
+3
+2
+1
-1
the reference matrix used by the gene finding methods is very important
Heterogeneity in genomis sequences
+3
+2
Acinetobacter
«natifs» gene model
+1
-1
-2
-3
The matrix used does not fit the codon usage of the
genes founded in this part of the sequence
Horizontal transfer ?
• start codon assertion (non-ATG / alternatifs)
Several existing problems • small genes detection
• « atypical » genes
AMIGene (S. Bocs)
Building one or more gene models : AMIMat
Annotation of
MIcrobial
Genes
Gene prediction using Markov Model
(Such as GeneMark)
Heuristic for the selection of the most
probable CDSs.
AMIGene et les modèles de gènes …
http://www.genoscope.cns.fr/agc/tools/amigene
Construction d’un modèle de
gènes à partir de la séquence
utilisateur (> 10 kb)
Utilisation des modèles de gènes
calculés pour un ensemble de
génomes (environ 80)
Gene model construction : AMIMat strategy
S. Cruveiller
presentation
Annotation fonctionnelle
« FONCTION » ?
• rôle biochimique
• rôle physiologique
• mécanisme
• expérimental
(gène rapporteur; expression différentielle...)
• par similarité de séquence
(criblage de banques)
• par contexte
(voisinage)
• « synténies »
• métabolisme
.…
?
Functional annotation tools
BlastP : Similarities searches in protein databanks and alignments
Also used for orthologs and paralogs identification
InterProScan : Searching for functionnal domains in Prosite,
PFAM, PRODOM databanks
D. Vallenet
presentation
Cognitor : Finding similarities in the Cluster of Orthologous Genes
(COG classification)
PRIAM : Finding similarities
L. Labarre
with enzymatic profiles
(enzymatic classification)
presentation
Pathway tools (BioCyc/P; Karp) : Metabolic pathway reconstruction
Syntonizer : Synteny group detection
SignalP /TMhmm : Peptide signal and Transmembrane helix predictions
AutoFAssign : Automatic functionnal assignation
From different authors
From the AGC group
Recherche de similarités : banques de protéines
+
CDSs traduites
= protéome
SWISSALL
BlastP
FastA
Pour une séquence peptidique
comparée, liste des protéines
des banques les plus “similaires”
(= hits blast).
• On opère un transfert par similarité de la fonction biologique présumée
(identité > 50% sur une longueur de 80% des séquences).
• On va propager des annotations du type ‘putative kinase’ à d’autres
protéines, ressemblant de moins en moins à la première.
=> quel est le seuil de ressemblance à partir duquel 2 protéines peuvent
avoir la même fonction ?
• Similarité en séquence/similarité en structure ou de la fonction
• annotations des banques incomplètes/fausses
=> propagation les erreurs d’annotation
• “Orphelins”
Recherche de similartiés : banques de motifs protéiques
Objectif : tenir compte de la modularité des protéines
+
CDSs traduites
= protéome
Banque de Programme
domaines
“ad-hoc”
protéiques
Pour une séquence peptidique,
caractéristiques des motifs
protéiques
les plus probables
• Domaines répertoriés sous forme de “profiles”
• Autant de programmes de recherche que de banques (formats
différents)
-> PROSITE, BLOCKS, PRINTS, PFAM, etc.
• Compléments des résultats de BlastP
=> éviter une annotation unique dans le cas de protéines
modulaires.
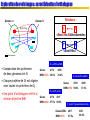
Exploration des voisinages : caractérisation d’orthologues
Genome A
Relations :
Genome B
Dyn. Prog.
1
1
1
«Best Hits Bidirectionnels»
1
1
1
n
1
2
3
2
«Best Hits»
Gène orphelin
E. coli/B. subtilis
• Comparaison des protéomes
de deux génomes A et B.
Genes
4174
BHB=1503 36.0%
4098
35.0%
• Chaque protéine de Gi est alignée
Genes
avec toutes les protéines de Gj.
• Une paire d’orthologues vérifie la
relation bijective BHB
S. aureus/B. subtilis
2593
BHB=1552 59.8%
4098
37.9%
E. coli/Y. pestis
Genes
4174
4017
BHB=2402 57.5% 59.8%
Y. pestis/Y.pseudotuberculosis
Genes/CDSs
4017
BHB=3518
87.6%
4347
80.9%
Groupes de Gènes Orthologues = COG (Koonin)
http://www.ncbi.nlm.nih.gov/COG/
Un COG = ens. de protéines qui devraient dériver d’une protéine ancestrale commune
Principe : • comparaisons 2 à 2 des protéomes de 70 génomes bactériens
• regroupement des gènes orthologues (BBH) : forment une classe
fonctionnelle particulière
PkGDB : Procaryotic Genome DataBase
Objectif : données d’annotation ‘propres’, cohérentes, à la source des
méthodologies de génomique comparative
• SGBD relationnel (MySQL)
• Génomes complets (Refseq NCBI)
Intégration dans PkGDB
Homogénéité des données
Gestion des ‘frameshifts’
Processus d’intégration des données publiques dans PkGDB
PkGDB
Fichiers
des
banques
PkGDB
Databank_Annotation
Données issues des
banques
Databank_Annotation
Données issues des
banques
Compare_Annotation
Ens. des CDSs ‘valides’
CDSs ‘valides’ des
banques (1)
Toutes les CDSs :
Jeu de CDSs (1)
+
CDSs dont les bornes ont été
corrigées automatiquement
OU à corriger manuellement
Construction des pré-matrices
(probabilités de transition/
modèle markovien)
Courbes de probabilité de
codage
• Correction/vérification des CDS à ‘problème’
• Annotation des pseudogènes
Exemple de corrections : annotation des pseudogènes
gene
Error type = ‘No3multiple’
gene
CDS
kdpC
kdpB
kdpD
kdpE
speF
gene
CDS ‘complexe’ (type cCDS)
gene
gene
CDSs ‘fragment’ (type fCDS)
…
622524..624571
/gene="kdpB"
/locus_tag="S0610"
/note="frameshift"
/pseudo
/db_xref="GeneID:1077039"
624580..625152
/gene="kdpC"
/locus_tag="S0611"
624580..625152
/gene="kdpC"
/locus_tag="S0611"
/function="enzyme; Transport of
small molecules: Cations"
/codon_start=1
/transl_table=11
/product="potassium-transporting
ATPase"
625145..627825
/gene="kdpD"
/locus_tag="S0612"
/note="frameshift"
/pseudo
627822..628507
/gene="kdpE"
/locus_tag="S0613"
/note="frameshift"
/pseudo
629197..631394
/gene="speF"
/locus_tag="S0614"
/note="frameshift"
/pseudo
Processus d’intégration des données publiques dans PkGDB
PkGDB
Fichiers
des
banques
PkGDB
Databank_Annotation
Données issues des
banques
Databank_Annotation
Données issues des
banques
Compare_Annotation
Databank_Annotation
Données issues des
banques
Compare_Annotation
Annotations banques
Statut = ‘Checked’
Ens. des CDSs ‘valides’
CDSs ‘valides’ des
banques (1)
PkGDB
Toutes les CDSs :
Jeu de CDSs (1)
+
CDSs dont les bornes ont été
corrigées automatiquement
OU à corriger manuellement
CDSs
corrigées/validées (2)
Construction des pré-matrices
(probabilités de transition/
modèle markovien)
Courbes de probabilité de
codage
AMIMat :
construction des
modèles de gènes
• Correction/vérification des CDS à ‘problème’
• Annotation des pseudogènes
PkGDB : Procaryotic Genome DataBase
Objectif : données d’annotation ‘propres’, cohérentes, à la source des
méthodologies de génomique comparative
• SGBD relationnel (MySQL)
• Génomes complets (Refseq NCBI)
Intégration dans PkGDB
Homogénéité des données
Gestion des ‘frameshifts’
Ré-annotation syntaxique
Complétion /correction des données
MICheck : ré-annotation (syntaxique) de génomes bactériens
Objectif : Vérifier rapidement si les annotations répertoriées dans les
banques de séquences pour un génome donné sont complètes.
Fichier EMBL ou GenBank
Séq. nucl
Annotations
+
Modèle(s) de gènes
CDS prédites
CDS UNIQUES
AMIGene
COMPARAISON
Position des codons stop
CDS communes
Calcul de la probabilité
moyenne de codage
Gènes annotés
CDS UNIQUES
Banques
http://www.genoscope.cns.fr/agc/tools/micheck
Cruveiller et al. (2005) MICheck : A Web tool to fast check annotations of bacterial genomes. Nucleic Acid Research (en révision)
Projets de ré-annotation de génomes bactériens
Base de données CMR (Comprehensive Microbial Resource) du TIGR
+
«Primary annotation» : annotations originales
Gènes en plus
« TIGR annotation » : annotations automatiques
(disponibles en consultation uniquement)
Les banques de séquences publiques
NCBI (Genbank) : projet Refseq (Reference Sequence)
Reviewed RefSeq : annotations automatiques + ‘curation’ manuelle par des
experts du NCBI.
Provisional RefSeq : annotations automatiques uniquement
Provisional RefSeq : annotations originales
Gènes en plus/en moins
Résultats MICheck sur A. pernix (status Reviewed Refseq)
CDS UNIQUES
AMIGene
18
35
CDS communes
1565
1569
CDS UNIQUES
Banques
941
186
BA000002
NC_000854
Genbank
‘original’
(BA000002)
Fichier
‘Refseq’
(NC_000854)
APE1077
APE1089
APE1087a
rplX
APE1088a
APE1097
Résultats MICheck sur O. iheyensis (status Reviewed Refseq)
CDS UNIQUES
AMIGene
2
14
CDS UNIQUES
Banques
CDS communes
3406
3392
18
18
BA000028
NC_004193
Fichier
‘Refseq’
(NC_004193)
gene
CDS
gene
CDS
gene
CDS
complement(2047445..2047618) BA000028
/gene="OB2021"
complement(2047445..2047618)
/gene="OB2021"
/product="hypothetical protein"
2047725..2048765
/gene="OB2022"
2047725..2048765
/gene="OB2022"
/EC_number="3.5.1.28"
/product="N-acetylmuramoyl-L-alanine
amidase (partial) "
/translation="MKLTTLISTIL… "
complement(2048799..2049245)
/gene="OB2023"
complement(2048799..2049245)
/gene="OB2023"
gene
complement(2047445..2047618)
NC_004193
/locus_tag="OB2021"
/db_xref="GeneID:1018510"
CDS
complement(2047445..2047618)
/locus_tag="OB2021"
/product="hypothetical protein"
misc_feature 2047725..2048765
/note="similar to N-acetylmuramoyl-L-alanine amidase"
gene
complement(2048799..2049245)
/locus_tag="OB2023"
/db_xref="GeneID:1018512"
CDS
complement(2048799..2049245)
/locus_tag="OB2023"
/note="CDS_ID OB2023
Projets de ré-annotation de génomes bactériens
Base de données CMR (Comprehensive Microbial Resource) du TIGR
+
«Primary annotation» : annotations originales
Gènes en plus
« TIGR annotation » : annotations automatiques
(disponibles en consultation uniquement)
Les banques de séquences publiques
NCBI (Genbank) : projet Refseq (Reference Sequence)
Reviewed RefSeq : annotations automatiques + ‘curation’ manuelle par des
experts du NCBI.
Provisional RefSeq : annotations automatiques uniquement
Provisional RefSeq : annotation originales
Gènes en plus/en moins
EBI (EMBL) : projet Genome Reviews
Enrichissement/correction des annotations fonctionnelles originales
(Données UniProt, Genome Ontology, InterPro, etc)
Standardisation/homogénéisation des annotations originales
Détection et élimination des annotations ‘erronées’ (système Xanthippe)
Gènes en moins
Résultats MICheck sur S. oneidensis (status Reviewed Refseq)
CDS UNIQUES
AMIGene
CDS communes
20
150
4114
4144
CDS UNIQUES
Banques
216
0
AE005176
AE005176_GR
Genbank
‘original’
(AE005176)
Fichier
Genome
Review
(AE005176_GR)
Fichier d’annotation original et fichier EMBL (GR)
gene
gene
CDS
gene
gene
CDS
gene
…
3266258..3268062
AE005176
/gene="dctB"
/locus_tag="SO3137"
/note="This region contains an authentic frame shift and
/note="This region contains an authentic frame shift and
is not the result of a sequencing artifact;
is not the result oftransport
a sequencing
artifact;
C4-dicarboxylate
C4-dicarboxylate
sensor
protein,
authentic
transport
sensor
protein,
authentic
frameshift"
frameshift"
3268059..3269438
/gene="dctD"
/locus_tag="SO3138"
3268059..3269438
/gene="dctD"
/locus_tag="SO3138"
/note="similar to GB:X14046, SP:P11049, and PID:29794;
identified by sequence similarity; putative"
/codon_start=1
/transl_table=11
/product="C4-dicarboxylate transport transcriptional
regulatory protein"
complement(3269514..3272585)
/locus_tag="SO3139"
/note="This region contains an authentic frame shift and
/note=" This region contains an authentic frame shift and
is not the result of a sequencing artifact; conserved
is not the result
of a sequencing
artifact;
… " putative"
hypothetical
protein;
identified by
Glimmer2;
complement(3273023..3273601)
/locus_tag="SO3140"
complement(3273023..3273601)
/locus_tag="SO3140"
/note="identified by match to PFAM protein family HMM
PF00265"
/codon_start=1
/transl_table=11
/protein_id="AAN56142.1"
/product="thymidine kinase
3274138..3276066
/locus_tag="SO3141"
/note="This region
region contains
contains aa gene
gene with
with one
one or
or more
more
/note="This
premature
stops
or
frameshifts,
and
is
not
the
result
of aa
premature stops or frameshifts, and is not the result of
sequencing
artifact;
cytochrome
c,
degenerate;
similar
to
sequencing artifact; cytochrome c, degenerate; similar to
GP:3628769;
identified
by
sequence
similarity;
putative"
GP:3628769; identified by sequence similarity; putative"
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
FT
CDS
CDS
CDS
CDS
3264761..3266158
AE005176_GR
/codon_start=1
/gene="dctM {UniProt/TrEMBL:Q8ECK2}"
/locus_tag="SO3136 {UniProt/TrEMBL:Q8ECK2}"
/product="C4-dicarboxylate transport protein …
3268059..3269438
/codon_start=1
/gene="dctD {UniProt/TrEMBL:Q8ECK1}"
/locus_tag="SO3138 {UniProt/TrEMBL:Q8ECK1}"
/product="C4-dicarboxylate transport
transcriptional regulatory protein
{UniProt/TrEMBL:Q8ECK1} »
complement(3273023..3273601)
/codon_start=1
/gene="tdk {UniProt/Swiss-Prot:Q8ECK0}"
/locus_tag="SO3140 {UniProt/SwissProt:Q8ECK0}"
/product="Thymidine kinase {UniProt/SwissProt:Q8ECK0}"
/EC_number="2.7.1.21 {UniProt/Swiss-Prot:Q8…}"
/function="ATP binding {GO:0005524} »
/function="thymidine kinase activity {GO:0004797}"
/biological_process="DNA metabolism
{GO:0006259}"
3276288..3278438
/codon_start=1
/gene="dcp-1 {UniProt/TrEMBL:Q8ECJ9}"
/locus_tag="SO3142 {UniProt/TrEMBL:Q8ECJ9}"
/product="Peptidyl-dipeptidase Dcp"
/function="metalloendopeptidase activity
{GO:0004222}"
/biological_process="proteolysis and peptidolysis
{GO:0006508}"
PkGDB : Procaryotic Genome DataBase
Objectif : données d’annotation ‘propres’, cohérentes, à la source des
méthodologies de génomique comparative
• SGBD relationnel (MySQL)
• Génomes complets (Refseq NCBI)
Intégration dans PkGDB
Homogénéité des données
Gestion des ‘frameshifts’
Ré-annotation syntaxique
Complétion /correction des données
• Génomes nouveaux (projets d’annotation)
• Résultats d’analyses :
Intrinsèques : gènes, signaux, répétitions,…
Extrinsèques : Blast, InterPro, COG, synténies
…
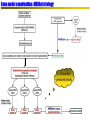
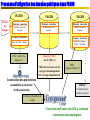
Stratégie générale de l’annotation des génomes bactériens -1Séquençage
Biological databases
Prédiction
automatique de gènes
Annotation
fonctionnelle (auto)
Annotation
manuelle
Prediction of coding regions,
promoters, terminators, RNAs
Similarity searches, assignments to
protein families, sequence features, …
Suggestion of function, classification
Validation of automatic annotations,
Additional database and literature searches,
Contextual analysis, gene fusions, protein
interactions,
Phylogenetic profiles
Intégration dans d’autres
plateformes d’analyse
Ré-annotation
Validation and update of previous annotations
Expression data, knock-out phenotypes, etc.
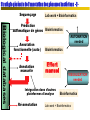
Stratégie générale de l’annotation des génomes bactériens -2Sequençage
Lab work + Bioinformatics
Biological databases
Prédiction
automatique de gènes Bioinformatics
Annotation
fonctionnelle (auto)
Annotation
manuelle
AUTOMATION
needed
Bioinformatics
Effort
manuel
Intégration dans d’autres
plateformes d’analyse
Ré-annotation
VISUALIZATION
needed
Bioinformatics
Lab work + Bioinformatics
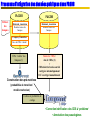
Schéma général du système MaGe
Specialized databases
Public databanks
«Private» sequences
PkGDB
MySQL
DB
tRNAscan-SE
Yersinia
Scope
Bacillus
Scope
ColiScope
Blast
PRIAM
InterProScan
COGnitor
TMHMM
FrankiaDB
AcinetoDB
HaloplanktisDB
«AutoFunc»
Automatic functional assignment
combining multiple evidence and
synteny results
Databases for annotation and
re-annotation projects
GRAPHICAL ANNOTATION INTERFACE
(Web server connected to the data base)
Validation and completion of the automatic annotation
(Re) Annotation using synteny results
Module d’assignation fonctionnelle automatique (AutoFunc) -1/label
CDS name (very different from gene name !) = CENARnumber
Genomes de Référence : E. coli et Acinetobacter ADP1
IF identity > 40% AND alignment on 80% of the protein lengths
OR identity > 30% AND alignment on 80% of the protein lengths AND SYNTENY
DA = « Definitive_Annotation »
/product
Description of the best hit : DA_SWALL
OR the one of Monica R. (EcoGene database) IF one E. coli protein is
similar to the annotated gene : DA_COLI
/gene
Gene name and synonyms from the EcoGene database IF one E. coli
protein is similar to the annotated gene.
/function
Functionnal Classification (E. coli)
/EC_number
PRIAM EC number(s)
IF identity > 40% AND partial alignment
Query protein
PM = Partial_Match
(>80% of the databank protein length)
/product
Databank protein
Description of the best hit : PM_SWALL
OR the one of Monica R. (EcoGene database) IF one E. coli protein is
similar to the annotated gene : PM_COLI
+ (partial match)
Module d’assignation fonctionnelle automatique (AutoFunc) -2IF identity > 40% AND partial alignment
FO= Fragment_Of
Query protein
(>80% of the query protein lenght)
Databank protein
Description of the best hit : PM_SWALL
OR the one of Monica R. (EcoGene database) IF one E. coli protein is
similar to the annotated gene : PM_COLI
+ (partial)
/product
IF 30% < identity < 40% AND alignment on 80% of the protein lengths
PA = Putative_Annotation
/product
Putative/Probable (?) + description of the best hit
PA_SWALL OR the one of E. coli PA_COLI
IF identity < 30% : no significant databank similarity
/product
/note
HP = Hypothetical_Protein
Hypothetical protein / Orphan Protein ?
Summary of the 3 SWALL best hits
Annotation définitive : example
2.1.1: DNA replication
Annotation définitive, partial match : example
Ratio of alignment lengths with Lmatch (length of match), Lprot1 (length of protein 1) and Lprot2 (length of protein 2)
minL = Lmatch/ min(Lprot1, Lprot2) and maxL = Lmatch /max(Lprot1, Lprot2)
Visualisation sous MaGe de CENAR0426
CENAR0426
Annotation définitive, partial : example
Visualisation sous MaGe de CENAR0361
CENAR0361
Erreur de séquence probable -> il manque le début du gène
(mettre CENAR361 à CheckSeq)
« Partial » and « partial match » : other cases
CENAR3153
3150
CENAR3149
3151
« partial
match»
« partial »
mdoH
mdoH mdoG
CENAR3149/3950 : « CheckSeq »
CENAR3153/56 : Ajuster le codon start
CENAR3156