Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Pharmaceutical marketing wikipedia , lookup
Polysubstance dependence wikipedia , lookup
Orphan drug wikipedia , lookup
Plateau principle wikipedia , lookup
Compounding wikipedia , lookup
Neuropsychopharmacology wikipedia , lookup
Psychopharmacology wikipedia , lookup
Neuropharmacology wikipedia , lookup
Drug design wikipedia , lookup
Pharmacognosy wikipedia , lookup
Drug discovery wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Pharmaceutical industry wikipedia , lookup
Prescription costs wikipedia , lookup
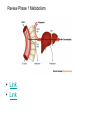
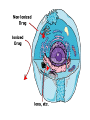
Double-blind Clinical Trials • A double-blind or double-masked study is one in which neither the participants nor the study staff know which participants are receiving the experimental treatment and which ones are receiving either a standard treatment or a placebo. • These studies are performed so that neither the patients’ nor the doctors’ expectations about the experimental drug can influence the outcome. Review Phase 1 Metabolism • Link • Link Is a Drug Polar or Non-polar (and why does this matter?) •To reach its target, the drug must pass through several membranes • If orally administered, this begins with the stomach and continues to the small and large intestine. “Like Dissolves Like” • To get across most membranes, the drug must be relatively non polar • To be soluble in water, a drug must be polar • If a drug is too nonpolar, it may be not be water soluble, or may bind too tightly to components in food, or to proteins in the blood. The polarity of a substance is measured by its partition coefficient in a two phase system consisting of 1octanol and water • P= [amount of drug dissolved in octanol] [amount of drug dissolved in water] • Usually the logarithm logP, is used to describe this ratio. • Christopher Lipinski noticed that most of the orally bioavailable drugs on the market seemed to have logP values less than 5. • There are now computer programs that will attempt to calculate this number from the structure. This calculated version is usually referred to as clogP, meaning calculated logP On the x-axis is plotted logP, and on the y-axis is plotted the permeability coefficient of rat brain capillaries in cm/sec. Note that, in general, more lipophilic compounds penetrate brain more rapidly. But some drugs change their ionic form, depending on the pH of the surrounding medium. Ionized (I.e. charged) states of molecules are always more polar than the uncharged forms. Two such classes of drugs are amines, R-NH2, and Carboxylic acids, RCOOH. At approximately pH = 12, the equilibrium below is evenly distributed between ammonium salt and amine. H H + N R H H2O R H + N HO H At the pH of blood, pH = 7.4, the equilibrium below is strongly shifted toward the ammonium salt. H H + N R H H H2O N R H + HO This is NOT true for amides RCONH2, Which are significantly different electronically from amines. Amides are Much harder to protonate. At pH = 7.4, amides exist in the unprotonated state, as shown. H H + N R C O H H2O H X R N C O H + HO Carboxylic acids are evenly distributed between charged, and uncharged form at pH = 4 O O + C R H2O OH + C R H3O O At pH = 7.4, the equilibrium lies in favor of the charged form. O O + C R OH H2O + C R O H3O HO- Group is needed for activity 2 HO 3 1 11 4 13 5 HO 10 15 12 O 9 14 H 8 6 Basic Nitrogen of Drug 16 H N CH3 7 Morphine (Astramorph) HO- Group not important to activity Lots of drugs have amines (primary, secondary, and tertiary) as a part of their structure. This allows the drug to exist in two forms, a charged version, which dissolves readily in water… As well as an uncharged form, which can easily cross membranes. • pH stomach = 1 to 3 (the stomach itself is protected by a layer of mucous). • pH small intestine = 8 • pH blood = 7.4 • Thus each drug will exist in different ionic states in different regions of the body. http://soolin.sunderland.ac.uk/fdcps/pharmacokinetics.ht ml Ways to administer a drug • Enteral = Through or within the intestines or gastrointestinal tract. • Parenteral = Not in or through the digestive system. Oral Administration • Easiest • Disadvantages – Some drugs (eg proteins) are not stable to the acidic environment and digestive enzymes of the stomach – May cause emesis – Drug may not be absorbed properly • Sublingual: Under the tongue. • Example: Nitroglycerin (brand name: nitrostat) • This medication is a nitrate used to relieve and prevent chest pain (angina) that occurs when the heart is deprived of oxygen • Nitroglycerin relaxes blood vessels allowing more blood to flow through. This reduces the workload on the heart and improves blood flow to the heart. Suppositories • Rectal: the substance crosses the rectal mucosa into the bloodstream • Vaginal: commonly used to treat gynaecological ailments, including vaginal infections such as candidiasis. Transdermal • Matrifen Fentanyl Patch Parenteral Routes • Intravascular (IV, IA)- placing a drug directly into the blood stream • Intramuscular (IM) - drug injected into skeletal muscle • Subcutaneous - Absorption of drugs from the subcutaneous tissues • Inhalation - Absorption through the lungs •Intraosseous infusion is the process of injection directly into the marrow of the bone. The needle is injected through the bone's hard cortex and into the soft marrow interior. • When IV access cannot be obtained in pediatric emergencies, intraosseous access is usually the next approach. It can be maintained for 24-48 hours, after which another route of access should be obtained. Intraosseous access is used less frequently in adult cases due to greater difficulty penetrating denser adult bone. Intrathecal Injection •An intrathecal injection (often simply called "intrathecal") is an injection into the spinal canal (intrathecal space surrounding the spinal cord), as in a spinal anaesthesia or in chemotherapy or pain management applications. Intrathecal Injection • This route is also used for some infections, particularly post-neurosurgical. The drug needs to be given this way to avoid the blood brain barrier. If the drug were given via other routes of administration where it would enter the blood stream it would be unable to reach the brain. • Drugs given intrathecally often have to be made up specially by a pharmacist or technician because they cannot contain any preservative or other potentially harmful inactive ingredients that are sometimes found in standard injectable drug preparations. Metabolism Link Link Pharmacokinetics and Pharmacodynamics Pharmacokinetics • • • • • • Defined as what the body does to the drug: Absorption Distribution Metabolism Excretion Pharmacokinetics uses mathematical models to predict the time-course of drug concentration in body fluids. Goal of Therapeutics • Achieve efficacy without toxicity • Plasma concentration (Cp) must be within the ‘therapeutic window’ • Cp units are mg/L • That is, it must be above the minimum effective concentration (MEC), and below the minimum toxic concentration (MTC) Fundamental Equations • • • • Cp = (dose rate)/Cl Dose rate has units mg/h Cp has units mg/L Cl = clearance (units are L/h), representing the volume cleared of drug per unit time • Link • Low clearance may be due to renal impairment, liver impairment, enzyme inhibition, age (old age or neonate). • Link Drug Clearance • To a first approximation, drugs are cleared from plasma in two ways, by metabolism in the liver and by being eliminated (unchanged) through the kidneys. • The fraction unchanged (fu) represents the proportion cleared by kidneys, while 1-fu represents the fraction cleared by metabolism. • Depending on the structure of the drug the proportion eliminated metabolically versus that eliminated renally will change. • Thus dosage must be adjusted to accommodate these factors. Link Volume of Distribution • However, drugs are distributed throughout the body, not just in plasma • Thus, as the drug spreads throughout the body, the plasma concentration falls, while maintaining an equilibrium concentration with other compartments • Ab = (Vd)(Cp) • Ab = total amount of drug in body (Amount in body, milligrams) • Vd = volume of distribution (liters) • Cp = plasma concentration (milligrams/liter) • Link The Half-Life of the Drug • The half-life of a drug is the amount of time required to reduce the concentration by 50% • The larger the volume of distribution, the longer it takes to clear the drug, at a constant rate of clearance. • t1/2 = (0.693)Vd/Cl • 0.693 = ln2 • Link Dosing Forms and Techniques • Oral availability is less than by IV • F = AUCpo/AUCIV • F = fraction of the drug given orally that reaches systemic circulation • AUCpo is the area under the concentration-time curve for the drug given orally (po) • AUCIV is the area under the concentration-time curve for the drug given by IV • ‘Loading Doses’ are larger than normal doses given at the beginning of treatment to rapidly increase Cp. • Link Oral Availability and Metabolism • Oral availability depends on both absorption and first pass metabolism • First pass metabolism can occur both in the liver and also in the gut wall. • Link Pharmacodynamics • Pharmacodynamics is defined as what the drug does to the body • Pharmacodynamics refers to the time-course and intensity of drug action and response. Pharmacodynamics • The potency of a drug is defined as the concentration need to achieve its maximum effect. It is often measured as EC50, the concentration required to achieve 50% of the maximum effect • The efficacy of a drug is defined as the absolute value of the maximum effect (Emax) (e.g. morphine is more efficacious as a pain reliever than acetaminophen) • Link Therapeutic Index • The therapeutic index represents the ratio of the concentration required to cause an adverse effect to the that required for the desired effect. • Therapeutic Index = EC50 (adverse effect) / EC50 (desired effect) • Pharmaceutical companies prefer drugs with a large therapeutic index. • Link Pharmacogentics • Among a population, different genotypes may result in different phenotypes that have different expression of receptors, drug metabolizing enzymes, or transporters, thus resulting in different susceptibility to a drug. • For a metabolizing enzyme, for example, one abberant allele can result in an intermediate metabolizer, while two abberant alleles may result in a poor metabolizer. Link • Examples include individuals of Asian descent who lack aldehyde dehydrogenase, thus do not tolerate alcohol and individuals who do not produce enough CYP2D6 in the liver to metabolize codeine to morphine and thus may not experience normal pain relief with this drug. Saturable Metabolism • A few drugs may saturate the enzymes responsible for their metabolism, thus resulting in higher than expected Cp. • Link Protein Binding of Drugs • Human serum albumin is the most abundant protein in human blood plasma • Acidic drugs, in particular, bind to serum albumin • The protein-bound form of the drug is unavailable to hit its target. • The protein-bound form of the drug must also dissociate from the protein in order to be cleared. pH and Pharmacokinetics • Acidic drugs usually contain weakly acidic functionalities, such as COOH. • Basic drugs usually contain weakly basic functionalities, such as amines. • Drugs which are acidic (pKa < 7), are ionized in basic media (pH > 7). • Drugs which are basic (pKa > 7) are ionized in acidic media (pH < 7) • The ionized form of the drug provides it with improved water solubility • But the unionized form generally passes nonpolar membranes more readily. • Link Dosing and Age • The dosing of drugs needs to be adjusted with the age of the patient. • Drug dosing may also need to be adjusted during pregnancy. Link Drug Interactions • Clearance can be altered by interaction with one or more drugs in a regimen • Enzyme inducers can serve to increase clearance and lower the plasma concentration of drugs. Examples include phenytoin, carbamazepin, and rifamycin. Drugs metabolized by CYP3A4 are particularly susceptible. • Enzyme inhibitors will decrease clearance and increase Cp. Examples include erythromycin, selective serotonin reuptake inhibitors (SSRIs), ketoconazole, amiodarone, cimetidine, grapefruit juice. • Link Drug Transporters • Specific transporters may aid influx, or alternatively, promote efflux of a drug. • One of the most important such systems is P-glycoprotein (permeability glycoprotein). • P-glycoprotein is a membrane-associate protein in the ATP binding cassette transporter superfamily (ABC transporter) P-Glycoprotein • P-glycoprotein can transport drugs back out of the gut wall and into the gut lumen, thus reducing absorption • It helps keep some drugs out of the brain • It transports drugs out of the kidney and into the urine. • P-glycoprotein has been implicated as a cause of multidrug resistance in tumor cells. • Link Reading Assignment: Goodman and Gilman’s Pharmaceutical Basis of Therapeutics, 12th edition, Chapter 2, pp. 17-39 Lin, Jiunn H.. Pharmacokinetic and pharmacodynamic variability: a daunting challenge in drug therapy. Current Drug Metabolism (2007), 8(2), 109-136. (assigned reading is only pp. 109-110, sections 1 and 2.0 and 129-132, sections 4-5). Link Raub, Thomas J. P-Glycoprotein Recognition of Substrates and Circumvention through Rational Drug Design. Molecular Pharmaceutics (2006), 3(1), 3-25 (assigned is pp. 3-9 and 24-25 only). Link Graduate Students Only: Goodman and Gilman’s Pharmaceutical Basis of Therapeutics, 12th edition, Chapter 6, pp. 123-143 Homework Questions 1) Mathematically define the following parameters, and their units: (Cl, Cp, Vd, Ab, fu, t1/2) 2) What is meant by the therapeutic index? 3) Explain what is meant by ‘targeted therapy’, using two examples of transtuzumab (Herceptin) and imatinib (Gleevec). 4) Why has the concept of ‘personalized medicine’ been so difficult to implement? 5) What is the best approach to designing a drug which is structurally optimized to elude P-gp?