Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
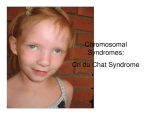
Dentistry Section DOI: 10.7860/JCDR/2013/4933.2988 Case Report The Crouzan Syndrome-A Case Report Manu Prasad, Ashwini S. Shetty, Manjula Shantaram ABSTRACT The Crouzon syndrome is a genetic disorder which is known as the brachial arch syndrome. It is an autosomal dominant disorder which is one of a rare group of syndromes which is characterized by cranio synostosis or a premature closing of the cranial sutures. The major features are brachiocephaly, occular proptosis, an under developed maxilla, mid face hypoplasia, a rare cleft lip or palate, hypodontia (some teeth missing) and crowding of teeth. Due to the maxillary hypoplasia, the Crouzon syndrome patients generally have a considerable permanent underbite and they subsequently cannot chew by using their incisors. We have presented in this article, a case of the Crouzon syndrome which was seen in a girl who was aged six years, with similar symptoms and the multidisciplinary approach which has to be followed in managing the case. Key Words: Craniofacial syndromes, Crouzon syndrome, Premature synostosis Introduction The Crouzon syndrome is named after the French neurologist, Octave Crouzon, who described this disorder [1-3] which includes a triad of skull deformities, facial anomalies, and an exopthalmus [4, 5]. Many bones which form the skull are separated by sutures which allow the skull to expand and develop in synchrony with the growth of the brain. Premature synostosis commonly involves the sagittal and the coronal sutures. Occasionally, the lambdoid sutures are involved. The order or rate of the suture fusion determines the degrees of deformity and disability [1, 2]. A premature sutural fusion may occur alone or together with other anomalies, thus causing various syndromes [6]. The craniofacial abnormalities are often present at birth and they may progress with time. A decreased mental function is present in approximately 12% of the patients. The family history may reveal individuals with a mild Crouzon syndrome. The headaches and a failing vision are attributable to an elevated intracranial pressure. In this article, we are presenting a case of the Crouzon syndrome which is one of the rare syndromes and is associated with synostosis of the cranial sutures, which is seen in 1 in 60,000 persons. The differential diagnosis of the Crouzon syndrome includes simple synostosis as well as the Apert, Pfeiffer and the Saethre- chotzen syndromes. ophthalmic features were, an extremely shallow orbit, an apparent proptosis, an inferior sclera which was caused by the maxillary hypoplasia, upper lid ptosis, nystagmus central (rotatory) exotropia and left eye myopia. The intercanthal distance was 28mm. There was a history of a spontaneous prolapse of the right eye ball, 2 to 3 years ago, due to rubbing of the eye. There was no recurrence. No digital abnormalities were present. [Table/Fig-1]: Picture showing exophthalmus Case report A six years old female child who was born by a full term normal delivery, with a normal appearance, developed an impaired vision with an excessive prominence of the eyeballs, an abnormal shape of the skull which was characterized by a premature fusion of the coronal sagittal and the bilateral parieto-occipital sutures, a maxillary complex (craniosynostosis), an exophthalmos, [Table/Fig-1] a beaked nose [Table/Fig-1], a short upper lip [Table/Fig-1], a hypoplastic maxilla, and a relative mandibular prognathism [Table/ Fig-2], crowding of the teeth, frontal bossing, a depressed nasal bridge, a broad nasal bone, low set ears, [Table/Fig-2] a high arched palate, a long philtrum and webbing of the neck [Table/Fig-3]. The Journal of Clinical and Diagnostic Research. 2013 May, Vol-7(5): 959-961 [Table/Fig-2]: Picture showing mandibular prognathism, frontal bossing, low set ears 959 Manu Prasad et al., The Crouzan Syndrome - A Case Report www.jcdr.net Discussion [Table/Fig-3]: Picture showing beaked nose, high arched palate, short upper lip, hypoplastic maxilla, long philtrum, webbing of the neck [Table/Fig-4]: Picture showing crowding of teeth, depressed nasal bridge and broad nasal bone The lack of sutural growth of the maxilla and the abnormal remodeling pattern led to a maxilla that was not only small in all the three dimensions but also, its internal architecture was deranged. The distance from the orbital floor to the nasal floor was extremely short and the distance from the nasal floor to the occlusal plane was increased. In the sagittal plane, the distance from the anterior nasal spine to the nasal spine was foreshortened. The mandible revealed a relatively normal growth pattern, whereby the patient grew into a class 3 relationship with the mandibular overjet, that is relative to a mandibular prognathism. An informed consent was obtained for publication. Investigations Blood: The peripheral blood smear showed a normocytic, hypochromic picture with mild leucopaenia CVS and RS: Normal X-ray report: Radiology of the skull revealed obliteration of the sagittal and the coronal sutures lines with an obvious bony continuity, mild ocular hypertelorism and crowding of the teeth. Features of the CT scan: A premature fusion of the multiple sutures was suggestive of craniosynostosis (fusion of the coronal sagittal and the bilateral parietooccipital sutures) and a mild hydrocephalus. Treatment A syndrome which is caused by the complexity of the symptoms always demands a multidisciplinary approach for a successful outcome. The aim of the treatment in this case, was to relieve the intracranial pressure, which can be done by creating a drainage of the CSF. This patient underwent a ventriculo peritoneal (VP) shunt for the mild hydrocephalus. 960 The phenotypic features of the Crouzon syndrome may be absent at birth and they may evolve gradually during the first few years of life [7, 8] as seen in the figures [Table/Fig-1-4]. It is commonly inherited as an autosomal dominant trait, with complete penetrance and a variable expressivity, but about one third of the cases do arise spontaneously. The male to female preponderance is 3:1 [2, 9]. With the advent of molecular technologies, the gene for the Crouzon syndrome could be localized to the Fibroblast Growth Factor Receptor 2nd gene (FGFR2), at the chromosomal locus 10q25.3q26, and more than 30 different mutations within the gene have been documented in separate families [10]. The cranial synostosis initiates changes in the brain and the adjoining structures such as an increased intracranial pressure, a reduced orbital volume, occlusal derangements and exophthalmoses [11]. A prenatal diagnosis of the exophthalmus has been reported by5 ultra sonography. So, an early recognition is essential, to guide the growth and development of the face and the cranium [7]. In 30% of the cases, the hydrocephalus is in progress, as per the literature. There was a mild dilatation of the ventricles in our patient too, which was suggestive of a hydrocephalus, which is one of the rare signs. A multidisciplinary approach was required to manage this case with treatment measures, to minimize the intracranial pressure and the secondary calvarias deformities. The innovations in craniofacial surgery enabled the patient to achieve their full potential, by maximizing their opportunities for an intellectual growth, a physical competence and a social acceptance. So, the prognosis depends on the severity of the malformations. The patients usually lead normal lives. The Crouzon syndrome requires major surgery and there can be major complications such as death or blindness, but in the hands of an experienced craniofacial team, this is rare. Other complications such as a collection of blood, an infection, bony irregularities, an electrolyte imbalance and a haemodynamical instability can occur. It should be remembered that these conditions are problems in the growth and that subsequent “re-corrective surgeries” may be required. References [1] Cohen MM Jr Craniosynostosis syndromes with craniosynostosis: Incidence, genetics penetrance, variability, and new updating. Birth Defects Orig Artic Ser. 1979;15: 13-63. [2] Cohen MM Jr. Craniosynostosis: Diagnosis, evaluation and management. Raven Press: New York; 1986. [3] Ahmed I, Afzal A. Diagnosis and evaluation of Crouzon syndrome. J Coll Physicians Surg Pak. 2009;19(5): 318-20. [4] Bowling EL, Burstein FD. Crouzon syndrome. Optometry. 2006; 77: 217-22. [5] Horbelt CV. Physical and oral characteristics of Crouzon syndrome, Apert syndrome, and Pierrerobin sequence. Gen Dent. 2008; 56(2):132-34. [6] Cohen MM Jr. Craniosynostosis update 1987. Am J Med Genet Suppl. 1988; 4: 99-148. [7] Atkinson FR. Hereditary craniofacial dysostosis or Crouzon’s syndrome disease. Med Press Circular. 1937;195: 118-24. [8] Golabi M. Radiographic abnormalities of Crouzan syndrome. A survey of 23 cases. Proc Greenwood Genet Ctr. 1984;3:102. [9] Gorlin RJ, Cohen MM, Levin LS. Syndromes of the head and neck. IN:,editors. 3rd ed. Oxford University Press: 1990; 516-26. [10] Jeftha A, StephenL, Morkel IA, Beighton P. Crouzono dermoskel etal syndrome. J Clin Pediatr Dent. 2004;28:173- 76. [11] Regezi JA, Sciubba JJ. Oral pathology-Clinical pathologic correlation. 3rd ed. WB Saunders Company: 1999; 436-37. Journal of Clinical and Diagnostic Research. 2013 May, Vol-7(5): 959-961 www.jcdr.net AUTHOR(S): 1. Dr. Manu Prasad 2. Dr. Ashwini S. Shetty 3. Dr. Manjula Shantaram PARTICULARS OF CONTRIBUTORS: 1. Consultant Surgeon, Department of Craniofacial Surgery, Yenepoya University Hospital, Yenepoya University, Mangalore 575 018, Karnataka, India. 2. Lecturer, Department of Anatomy, Yenepoya Medical College, Yenepoya University, Mangalore-575 018, Karnataka, India. 3. Associate Professor, Department of Biochemistry, Yenepoya Medical College, Yenepoya University, Mangalore 575 018, Karnataka, India. Journal of Clinical and Diagnostic Research. 2013 May, Vol-7(5): 959-961 Manu Prasad et al., The Crouzan Syndrome - A Case Report NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Manu Prasad, Department of Craniofacial Surgery Yenepoya University Hospital, Yenepoya University, Mangalore 575 018, Karnataka, India. E-mail: [email protected] Financial OR OTHER COMPETING INTERESTS: None. Date of Submission: Aug 08, 2012 Date of Peer Review: Oct 21, 2012 Date of Acceptance: Feb 14, 2013 Date of Publishing: May 01, 2013 961