Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
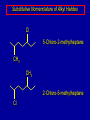
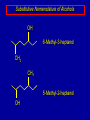
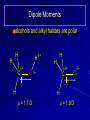
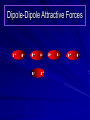
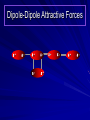
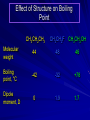
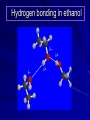
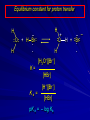
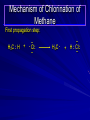
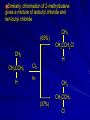
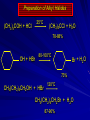
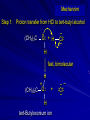
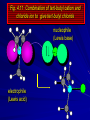
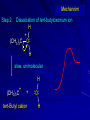
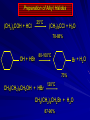
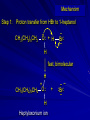
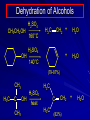
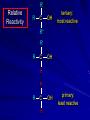
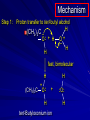
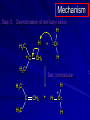
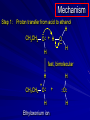
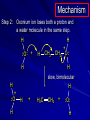
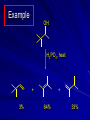
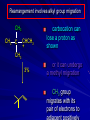
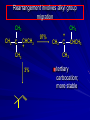
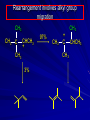
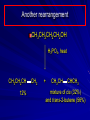
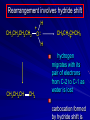
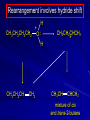
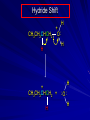
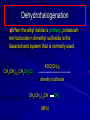
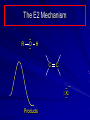
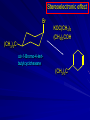
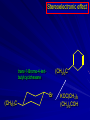
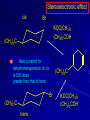
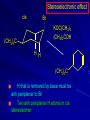
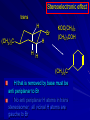
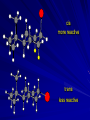
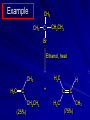
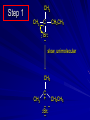
Alcohols and Alkyl Halides Overview of Chapter This chapter introduces chemical reactions and their mechanisms by focusing on two reactions that yield alkyl halides. (1) alcohol + hydrogen halide ROH + HX RX + H2O (2) alkane + halogen RH + X2 RX + HX Both are substitution reactions 4.1 IUPAC Nomenclature of Alkyl Halides IUPAC Nomenclature There are several kinds of IUPAC nomenclature. The two that are most widely used are: functional class nomenclature substitutive nomenclature Both types can be applied to alcohols and alkyl halides. Functional Class Nomenclature of Alkyl Halides Name the alkyl group and the halogen as separate words (alkyl + halide) CH3F CH3CH2CHCH2CH2CH3 Br CH3CH2CH2CH2CH2Cl H I Functional Class Nomenclature of Alkyl Halides Name the alkyl group and the halogen as separate words (alkyl + halide) CH3F CH3CH2CH2CH2CH2Cl Methyl fluoride Pentyl chloride CH3CH2CHCH2CH2CH3 Br 1-Ethylhexyl bromide H I Cyclohexyl iodide Substitutive Nomenclature of Alkyl Halides Name as halo-substituted alkanes. Number the longest chain containing the halogen in the direction that gives the lowest number to the substituted carbon. CH3CH2CH2CH2CH2F CH3CHCH2CH2CH3 Br CH3CH2CHCH2CH3 I Substitutive Nomenclature of Alkyl Halides Name as halo-substituted alkanes. Number the longest chain containing the halogen in the direction that gives the lowest number to the substituted carbon. CH3CH2CH2CH2CH2F 1-Fluoropentane CH3CH2CHCH2CH3 I 3-Iodopentane CH3CHCH2CH2CH3 Br 2-Bromopentane Substitutive Nomenclature of Alkyl Halides Cl CH3 CH3 Cl Halogen and alkyl groups are of equal rank when it comes to numbering the chain. Number the chain in the direction that gives the lowest number to the group (halogen or alkyl) that appears first. Substitutive Nomenclature of Alkyl Halides Cl 5-Chloro-2-methylheptane CH3 CH3 2-Chloro-5-methylheptane Cl 4.2 IUPAC Nomenclature of Alcohols Functional Class Nomenclature of Alcohols Name the alkyl group and add "alcohol" as a separate word. CH3CH2OH CH3 CH3CCH2CH2CH3 CH3CHCH2CH2CH2CH3 OH OH Functional Class Nomenclature of Alcohols Name the alkyl group and add "alcohol" as a separate word. CH3CH2OH Ethyl alcohol CH3CHCH2CH2CH2CH3 OH 1-Methylpentyl alcohol CH3 CH3CCH2CH2CH3 OH 1,1-Dimethylbutyl alcohol Substitutive Nomenclature of Alcohols Name as "alkanols." Replace -e ending of alkane name by -ol. Number chain in direction that gives lowest number to the carbon that bears the —OH group. CH3CH2OH CH3 CH3CCH2CH2CH3 CH3CHCH2CH2CH2CH3 OH OH Substitutive Nomenclature of Alcohols Name as "alkanols." Replace -e ending of alkane name by -ol. Number chain in direction that gives lowest number to the carbon that bears the —OH group. CH3CH2OH Ethanol CH3CHCH2CH2CH2CH3 OH 2-Hexanol CH3 CH3CCH2CH2CH3 OH 2-Methyl-2-pentanol Substitutive Nomenclature of Alcohols OH CH3 CH3 OH Hydroxyl groups outrank alkyl groups when it comes to numbering the chain. Number the chain in the direction that gives the lowest number to the carbon that bears the OH group Substitutive Nomenclature of Alcohols OH 6-Methyl-3-heptanol CH3 CH3 5-Methyl-2-heptanol OH 4.3 Classes of Alcohols and Alkyl Halides Classification Alcohols and alkyl halides are classified as primary secondary tertiary according to their "degree of substitution." Degree of substitution is determined by counting the number of carbon atoms directly attached to the carbon that bears the halogen or hydroxyl group. Classification H CH3CH2CH2CH2CH2F OH primary alkyl halide secondary alcohol CH3 CH3CHCH2CH2CH3 Br secondary alkyl halide CH3CCH2CH2CH3 OH tertiary alcohol Bonding in Alcohols and Alkyl Halides Dipole Moments alcohols and alkyl halides are polar H H H d+ C d+ H H d+ O d– H C Cl H m = 1.7 D m = 1.9 D d– Dipole Moments alcohols and alkyl halides are polar m = 1.7 D m = 1.9 D Dipole-Dipole Attractive Forces d+ d– d+ d– d– d+ d+ d– d+ d– Dipole-Dipole Attractive Forces d+ d– d+ d– d– d+ d+ d– d+ d– Physical Properties of Alcohols and Alkyl Halides: Intermolecular Forces Boiling point Solubility in water Density Effect of Structure on Boiling Point CH3CH2CH3 CH3CH2F CH3CH2OH Molecular weight 44 48 46 Boiling point, °C -42 -32 +78 0 1.9 1.7 Dipole moment, D CH3CH2CH3 Molecular weight 44 Intermolecular forces are weak. Boiling point, °C -42 Only intermolecular forces are induced dipole-induced dipole attractions. Dipole moment, D 0 CH3CH2F Molecular weight 48 Boiling point, °C -32 Dipole moment, D 1.9 A polar molecule; therefore dipole-dipole and dipole-induced dipole forces contribute to intermolecular attractions. CH3CH2OH Molecular weight 46 Boiling point, °C +78 Dipole moment, D 1.7 Highest boiling point; strongest intermolecular attractive forces. Hydrogen bonding is stronger than other dipole-dipole attractions. Hydrogen bonding in ethanol d– d– d+ d+ Hydrogen bonding in ethanol Boiling point increases with increasing number of halogens Compound CH3Cl CH2Cl2 CHCl3 CCl4 Boiling Point -24°C 40°C 61°C 77°C Even though CCl4 is the only compound in this list without a dipole moment, it has the highest boiling point. Induced dipole-induced dipole forces are greatest in CCl4 because it has the greatest number of Cl atoms. Cl is more polarizable than H. But trend is not followed when halogen is fluorine Compound CH3CH2F CH3CHF2 CH3CF3 CF3CF3 Boiling Point -32°C -25°C -47°C -78°C Fluorine is not very polarizable and induced dipoleinduced dipole forces decrease with increasing fluorine substitution. Solubility in water Alkyl halides are insoluble in water. Methanol, ethanol, isopropyl alcohol are completely miscible with water. The solubility of an alcohol in water decreases with increasing number of carbons (compound becomes more hydrocarbon-like). Hydrogen Bonding Between Ethanol and Water d+ d– d+ d+ d– d– Density Alkyl fluorides and alkyl chlorides are less dense than water. Alkyl bromides and alkyl iodides are more dense than water. All liquid alcohols have densities of about 0.8 g/mL. 4.6 Acids and Bases: General Principles Acids and Bases: General Principles Brønsted-Lowry definition an acid is a proton donor a base is a proton acceptor Proton Transfer from HBr to Water hydronium ion H .. O .. H .. . . H Br . . base acid H .. O+ H H .. . – .. Br . . . conjugate conjugate acid base Acid Strength A strong acid is one that is completely ionized in water. A weak acid is one that ionizes in water to the extent of less than 100%. Acid strength is measured by Ka or pKa Equilibrium constant for proton transfer H .. . – . .. O+ . H + . Br . . H H .. . .. O .. + H Br . . . H K= [H3O+][Br–] [HBr] [H+][Br–] Ka = [HBr] pK a = – log Ka Table 4.2 Strength of Some Brønsted Acids Acid Ka pKa Conj. Base 10 –10 I 9 –9 Br 7 –7 Cl –4.8 HSO4 –1.7 H 2O HI ~10 HBr ~10 HCl ~10 H2SO4 1.6 x 10 + H 3O 55.5 5 – – – – strong acids are stronger than hydronium ion Table 4.2 Strength of Some Brønsted Acids Acid H 3O + HF CH3CO2H + NH4 H2O Ka pKa Conj. Base 55.5 –1.7 H 2O 3.5 x 10 1.8 x 10 5.6 x 10 1.8 x 10 –4 –5 –10 –16 – 3.5 F 4.6 – CH3CO2 9.2 NH3 15.7 – HO weak acids are weaker than hydronium ion Table 4.2 Strength of Some Brønsted Acids Acid Ka H 2O 1.8 x 10 CH3OH ~10 CH3CH2OH ~10 (CH3)2CHOH ~10 (CH3)3COH ~10 pKa -16 Conj. Base – 15.7 HO -16 ~16 CH3O -16 ~16 CH3CH2O -17 ~17 (CH3)2CHO -18 ~18 (CH3)3CO – – – – alcohols resemble water in acidity; their conjugate bases are comparable to hydroxide ion in basicity Table 4.2 Strength of Some Brønsted Acids Acid NH3 (CH3)2NH Ka -36 ~10 -36 ~10 pKa ~36 ~36 Conj. Base NH2 – (CH3)2N – ammonia and amines are very weak acids; their conjugate bases are very strong bases Proton Transfer to Alcohols alkyloxonium ion R .. O .. H .. . . H Br . . base acid R .. O+ H H .. . – .. Br . . . conjugate conjugate acid base 4.7 Acid-Base Reactions: A Mechanism for Proton Transfer Proton Transfer from HBr to Water H .. O .. H .. . . H Br . . base acid H .. O+ H H .. . – .. Br . . . conjugate conjugate acid base Mechanism: single step transition state Potential energy activation energy reactants products Reaction coordinate Mechanism: single step d+ H2O Potential energy H2O + H—Br Reaction coordinate d- H Br bimolecular: both H2O and HBr involved at transition state + H2O—H + Br – Halogenation of Alkanes RH + X2 RX + HX Energetics RH + X2 RX + HX explosive for F2 exothermic for Cl2 and Br2 endothermic for I2 Chlorination of Methane carried out at high temperature (400 °C) CH4 + Cl2 CH3Cl + HCl CH3Cl + Cl2 CH2Cl2 + HCl CH2Cl2 + Cl2 CHCl3 + HCl CHCl3 + Cl2 CCl4 + HCl Mechanism of Chlorination of Methane Free-radical chain mechanism. Initiation step: .. .. .. .. . + . Cl: : Cl: Cl : : Cl .. .. .. .. The initiation step "gets the reaction going" by producing free radicals—chlorine atoms from chlorine molecules in this case. Initiation step is followed by propagation steps. Each propagation step consumes one free radical but generates another one. Mechanism of Chlorination of Methane First propagation step: H3C : H + .. . Cl: .. H3C . + .. H : Cl: .. First propagation step: H3C : H + .. . Cl: .. Second propagation step: .. .. : H3C . + : Cl: Cl .. .. H3C . + .. H : Cl: .. .. .. H3C: C : + . Cl: .. .. l First propagation step: H3C : H + .. . Cl: .. Second propagation step: .. .. : H3C . + : Cl: Cl .. .. .. .. : Cl : H3C : H + : Cl .. .. H3C . + .. H : Cl: .. .. .. H3C: C : + . Cl: .. .. l .. .. H3C: C : + H : Cl: .. .. l Almost all of the product is formed by repetitive cycles of the two propagation steps. First propagation step: H3C : H + .. . Cl: .. Second propagation step: .. .. : H3C . + : Cl: Cl .. .. .. .. : Cl : H3C : H + : Cl .. .. H3C . + .. H : Cl: .. .. .. H3C: C : + . Cl: .. .. l .. .. H3C: C : + H : Cl: .. .. l Termination Steps stop chain reaction by consuming free radicals .. H3C: C : .. l hardly any product is formed by termination step because concentration of free radicals at any instant is extremely low .. H3C . + . Cl: .. Chlorination of Alkanes can be used to prepare alkyl chlorides from alkanes in which all of the hydrogens are equivalent to one another CH3CH3 + Cl2 420°C CH3CH2Cl + HCl (78%) + Cl2 hn Cl + HCl (73%) Chlorination of Alkanes Major limitation: Chlorination gives every possible monochloride derived from original carbon skeleton. Not much difference in reactivity of different hydrogens in molecule. Example Chlorination of butane gives a mixture of 1-chlorobutane and 2-chlorobutane. (28%) CH3CH2CH2CH2Cl CH3CH2CH2CH3 Cl2 hn CH3CHCH2CH3 (72%) Cl Percentage of product that results from substitution of indicated hydrogen if every collision with chlorine atoms is productive 10% 10% 10% 10% 10% 10% 10% 10% 10% 10% Percentage of product that actually results from replacement of indicated hydrogen 18% 18% 4.6% 4.6% 4.6% 4.6% 4.6% 4.6% 18% 18% Relative rates of hydrogen atom abstraction divide by 4.6 18% 4.6% 4.6 =1 4.6 18 4.6 = 3.9 A secondary hydrogen is abstracted 3.9 times faster than a primary hydrogen by a chlorine atom. Similarly, chlorination of 2-methylbutane gives a mixture of isobutyl chloride and tert-butyl chloride (63%) CH3 CH3CCH2Cl CH3 CH3CCH3 H H Cl2 hn CH3 CH3CCH3 (37%) Cl Percentage of product that results from replacement of indicated hydrogen 7.0% 37% Relative rates of hydrogen atom abstraction divide by 7 7.0 7 =1 37 7 = 5.3 A tertiary hydrogen is abstracted 5.3 times faster than a primary hydrogen by a chlorine atom. Selectivity of free-radical halogenation R3CH > R2CH2 > RCH3 chlorination: 5 4 1 bromination: 1640 82 1 Chlorination of an alkane gives a mixture of every possible isomer having the same skeleton as the starting alkane. Useful for synthesis only when all hydrogens in a molecule are equivalent. Bromination is highly regioselective for substitution of tertiary hydrogens. Major synthetic application is in synthesis of tertiary alkyl bromides. Preparation of Alkyl Halides from Alcohols and Hydrogen Halides ROH + HX RX + H2O Reaction of Alcohols with Hydrogen Halides ROH + HX RX + HOH Hydrogen halide reactivity HI most reactive HBr HCl HF least reactive Reaction of Alcohols with Hydrogen Halides ROH + HX RX + HOH Alcohol reactivity R3COH Tertiary R2CHOH Secondary most reactive RCH2OH CH3OH Primary Methanol least reactive Preparation of Alkyl Halides 25°C (CH3)3COH + HCl (CH3)3CCl + H2O 78-88% OH + HBr 80-100°C Br + H2O 73% CH3(CH2)5CH2OH + HBr 120°C CH3(CH2)5CH2Br + H2O 87-90% Preparation of Alkyl Halides A mixture of sodium bromide and sulfuric acid may be used in place of HBr. CH3CH2CH2CH2OH NaBr H2SO4 heat CH3CH2CH2CH2Br 70-83% Mechanism of the Reaction of Alcohols with Hydrogen Halides Mechanism Step 1: Proton transfer from HCl to tert-butyl alcohol (CH3)3C .. O: + H .. : Cl .. H fast, bimolecular H + (CH3)3C O : + H tert-Butyloxonium ion .. – : Cl: .. Mechanism Step 2: Dissociation of tert-butyloxonium ion H + (CH3)3C O : H slow, unimolecular H + (CH3)3C + :O: tert-Butyl cation H Mechanism Step 3: Capture of tert-butyl cation by chloride ion. (CH3)3C + + .. – : Cl: .. fast, bimolecular (CH3)3C .. Cl .. : tert-Butyl chloride Carbocations + + (CH3)3C .. – : Cl: .. (CH3)3C .. Cl .. : The last step in the mechanism of the reaction of tert-butyl alcohol with hydrogen chloride is the reaction between an electrophile and a nucleophile. tert-Butyl cation is the electrophile. Chloride ion is the nucleophile. Fig. 4.11 Combination of tert-butyl cation and chloride ion to give tert-butyl chloride nucleophile (Lewis base) + electrophile (Lewis acid) – Potential Energy Diagrams for Multistep Reactions: The SN1 Mechanism Extension The potential energy diagram for a multistep mechanism is simply a collection of the potential energy diagrams for the individual steps. Consider the mechanism for the reaction of tert-butyl alcohol with HCl. (CH3)3COH + HCl 25°C (CH3)3CCl + H2O Mechanism Step 1: Proton transfer from HCl to tert-butyl alcohol (CH3)3C .. O: + H .. : Cl .. H fast, bimolecular H + (CH3)3C O : + H tert-butyloxonium ion .. – : Cl: .. Mechanism Step 2: Dissociation of tert-butyloxonium ion H + (CH3)3C O : H slow, unimolecular H + (CH3)3C + tert-Butyl cation :O: H Mechanism Step 3: Capture of tert-butyl cation by chloride ion. (CH3)3C + + .. – : Cl: .. fast, bimolecular (CH3)3C .. Cl .. : tert-Butyl chloride carbocation formation carbocation capture R+ proton transfer ROH + ROH2 RX carbocation formation d– d+ (CH3)3C O carbocation capture H Cl R+ H ROH + ROH2 RX H d+ (CH3)3C O d+ carbocation capture H R+ proton transfer ROH + ROH2 RX d+ carbocation formation (CH3)3C R+ proton transfer ROH + ROH2 RX d– Cl Mechanistic notation The mechanism just described is an example of an SN1 process. SN1 stands for substitution-nucleophilicunimolecular. The molecularity of the rate-determining step defines the molecularity of the overall reaction. Mechanistic notation The molecularity of the rate-determining step defines the molecularity of the overall reaction. H d+ (CH3)3C O d+ H Rate-determining step is unimolecular dissociation of alkyloxonium ion. 4.12 Effect of Alcohol Structure on Reaction Rate slow step is: ROH2+ R+ + H2O The more stable the carbocation, the faster it is formed. Tertiary carbocations are more stable than secondary, which are more stable than primary, which are more stable than methyl. Tertiary alcohols react faster than secondary, which react faster than primary, which react faster than methanol. Hammond's Postulate If two succeeding states (such as a transition state and an unstable intermediate) are similar in energy, they are similar in structure. Hammond's postulate permits us to infer the structure of something we can't study (transition state) from something we can study (reactive intermediate). carbocation formation carbocation capture R+ proton transfer ROH + ROH2 RX carbocation formation R+ proton transfer ROH + ROH2 Rate is carbocation governed by capture energy of this transition state. Infer structure of this transition state from structure of state of closest energy; in this case the nearest state is the RXcarbocation. Reaction of Primary Alcohols with Hydrogen Halides. The SN2 Mechanism Preparation of Alkyl Halides 25°C (CH3)3COH + HCl (CH3)3CCl + H2O 78-88% OH + HBr 80-100°C Br + H2O 73% CH3(CH2)5CH2OH + HBr 120°C CH3(CH2)5CH2Br + H2O 87-90% Preparation of Alkyl Halides Primary carbocations are too high in energy to allow SN1 mechanism. Yet, primary alcohols are converted to alkyl halides. Primary alcohols react by a mechanism called SN2 (substitution-nucleophilic-bimolecular). CH3(CH2)5CH2OH + HBr 120°C CH3(CH2)5CH2Br + H2O 87-90% The SN2 Mechanism Two-step mechanism for conversion of alcohols to alkyl halides: (1) proton transfer to alcohol to form alkyloxonium ion (2) bimolecular displacement of water from alkyloxonium ion by halide Example CH3(CH2)5CH2OH + HBr 120°C CH3(CH2)5CH2Br + H2O Mechanism Step 1: Proton transfer from HBr to 1-heptanol CH3(CH2)5CH2 .. O: + H .. : Br .. H fast, bimolecular H + CH3(CH2)5CH2 O : H Heptyloxonium ion + .. – : Br: .. Mechanism Step 2: Reaction of alkyloxonium ion with bromide ion. H .. – + : Br: + CH3(CH2)5CH2 O : .. H slow, bimolecular H CH3(CH2)5CH2 .. Br .. : 1-Bromoheptane + :O: H d+ d– Br CH2 OH2 CH3(CH2)4 CH2 proton transfer ROH + ROH2 RX Sostituzione nucleofila bimolecolare H3C + Br OH bromuro di metile H3C + Br metanolo CH3 H3C Br C2H5 OH + OCH3 H 2R 2-bromobutano H3CO + H C2H5 2S metossibutano Br Meccanismo della sostituzione nucleofila alifatica bimolecolare SN2 cinetica stereochimica struttura del substrato nucleofili gruppi uscenti solventi H3C Br + OH bromuro di metile v = k [CH3Br][-OH] H3C OH metanolo + Br CH3 H3C Br C2H5 H + OCH3 H3CO + H Br C2H5 2S 2R Inversione di Configurazione Reazione stereospecifica in quanto lo stereoisomero di partenza è trasformato in uno specifico stereoisomero del prodotto 110 Reattività relativa dei bromuri alchilici k rel CH 3 Br 150 CH 3 CH 2 Br 1 CH 3 CH Br 0 .0 0 8 CH 3 CH 3 CH 3 C CH 3 Br unreac t iv e! Nucleofilo KI R Cl + - OH R OH + - Cl R Cl + - R OH OH + - Cl R METILE ETILE ISOPROPILE T-BUTILE NEOPENTILE v 30 1 0,3 0 10-6 Gli stati di transizione delle reazioni spiegano la reattività differente in quanto un aumento dell’ingombro sterico al carbonio diminuisce la stabilità dello stato di transizione (maggiore energia) facendo aumentare l’Eatt e quindi diminuire la velocità. Nu L + nucleofilo prodotto OH alcoli H2O RO- alcoli ROH eteri - HS CN RC C NH3 I eteri tioli nitrili alchini ammine ioduri RCOO esteri RCOOH esteri d Nu d L +L Nu solventi costante dielettrica O diossano 2 O 2 C6H14 esano C6H6 benzene 3 C2H5OC2H5etere etilico 4 CH3COOH a. acetico NH3 ammoniaca 6 17 CH3CH2OH alcol etilico 24 CH3COCH3 acetone 25 HCOOH acido formico 59 H2O acqua 80 solventi polari aprotici H3C O dimetilsolfossido S O N C H H3C H C 38 3 H3C dimetilformammide 45 IDROSSIDO DI SODIO ACQUOSO CIANURO DI POTASSIO ORDINE DI NUCLEOFILICITA' elettroni elettroni non legame elettroni gli anioni sono più nucleofili degli acidi coniugati RO- ROH a parità di centro nucleofilo l'ordine di nucleofilicità corrisponde a quello di basicità ROHOper gli elementi di uno stesso gruppo la nucleofilicità aumenta con il peso atomico (in solventi protici) I- Br- Cl- F- H2Se > H2S > H2O, and PH3 > NH3 L’effetto è dovuto alla maggiore polarizzabilità degli atomi più grandi. Le nubi elettroniche più grandi permettono una maggiore sovrapposizione nello stato di transizione SN2. La nucleofilicità decresce andando da sinistra a destra nella tavola periodica H2N- > HO- > FI nucleofili stericamente ingombrati reagiscono più lentamente. I migliori gruppi uscenti sono le basi molto deboli Ioduro > bromuro > cloruro Lo ione idrossido è un pessimo gruppo uscente gruppo uscente NH2-, RO-, OH-, F-, Cl-, H2O, Br-, I-, CH3C6H4SO3-, CH3SO3O R O S CH3 R OTs O para-Toluenesulfonate Tosylate O R O S Br R OBs O para-Bromobenzenesulfonate Brosylate Quando un soluto viene sciolto in un solvente le molecole o gli ioni possono essere circondate da molecole del solvente ed essere pertanto solvatate. I solventi si distinguono in: solventi polari protici (formano legami idrogeno) acqua, alcoli solventi aprotici (non formano legami idrogeno) benzene, esano solventi polari aprotici (non formano legami idrogeno) ma hanno alta costante dielettrica ed alta polarità DMSO, DMF In generale la solvatazione formando una sfera di solvente intorno al nucleofilo indebolisce la sua forza attaccante un elettrofilo. Gli ioni più piccoli sono solvatati meglio dei più grandi. Così F- è maggiormente solvatato rispetto ad I-. Because aprotic solvents do not form hydrogen bonds, they solvate anionic nucleophiles relatively weakly. This results in an increase in the nucleophiles reactivity. Bromomethane reacts with KI 500 times faster in propanone than in methanol. Consider the reaction of iodomethane with chloride: The rate of the reaction is more than 106 times greater in the aprotic solvent DMF than in methanol. Scegliedo solventi aprotici la reattività aumenta ed ovviamente l’effetto è più marcato nel caso di piccoli anioni. Il bromometano reagisce con KI 500 volte più velocemente in acetone che in metanolo. Nella reazione dello iodometano con Cl- la velocità della Reazione passando dal metanolo alla DMF aumenta di 106 volte. Solvolisi del bromuro di t-butile La velocità delle reazioni SN2 diminuisce drasticamente quando il centro di reazione passa da primario a secondario e terziario. Questo è vero solo per le sostituzioni bimolecolari. Gli alogenuri secondari e terziari possono essere sostituiti con un meccanismo differente. Consideriamo la reazione del bromuro di t-butile ed acqua. (CH3)3CBr + H2O (CH3)3COH + HBr Quando un substrato subisce sostituzione ad opera del solvente la reazione prende il nome di solvolisi. L’acqua è il nucleofilo anche se il suo carattere nuclefilico è basso. La reazione non avviene con il bromuro di isopropile, il bromuro di etile e quello di metile. The rate law for the solvolysis of 2-bromo-2-methylpropane by water in formic acid (polar, low nucleophilicity) has been determined by varying the concentrations of the two reactants and measuring the rate of solvolysis. The rate law consistent with the kinetic data depends only on the halide concentration, not the water concentration: Rate = k[(CH3)3CBr] mol L-1 s-1 Two account for this kind of behavior it is necessary to postulate a mechanism consisting of 2 or more steps having an initial ratedetermining step, or slowest step which does not involve a water molecule. The sum of all of the steps in the proposed mechanism must add up to the observed overall reaction as shown in the stoichiometric equation. Sostituzione nucleofila monomolecolare CH3 CH3 CH3 C CH3 + acetone H2O CH3 C CH3 + H Br OH Br + other products v = k [alogenuro] (CH3)3CBr + H2O (CH3)3COH + HBr CH3Br CH3CH2Br (CH3)2CHBr (CH3)3CBr 0,001 0,01 (CH3)3COH + HBr 0,012 1200 (CH3)3CBr + H2O CH3OH < CH3CH2OH < (CH3)2CHOH < (CH3)3COH Br H C2H5 CH3 H3C H3C OCH3 H3CO + CH3OH H C2H5 H C2H5 1) CH3 CH3 C CH3 CH3 slow CH3 C + .. _ : Br : .. + CH3 : Br : .. 2) CH3 CH3 C + H CH3 + :O: CH3 fast CH3 C H :O CH3 + H H 3) CH3 CH3 fast CH3 C :O H CH3 + H CH3 C :O .. CH3 + H + H 1935: Hughes & Ingold 1) CH3 CH3 C 2) CH3 slow CH CH3 CH3 Br CH3 CH3 CH3 CH3 C CH3 C + CH CH3 + + Br CH CH3 CH3 CH3 CH3 C + CH CH3 + CH3 CH3 3) CH3 C _ CH CH3 CH3 + ROH CH3 C CH CH3 OR CH3 + + H C2H5 C2H5 C2H5 Cl + H2O OH HO H C CH3 H3C 3 C6H5 C6H5 C6H5 2-idrossi-2-fenilbutano (2R) 2-cloro-2-fenilbutano R 40% Nu Nu L S 60% Nu L L Nu Nu Nu Other Methods for Converting Alcohols to Alkyl Halides Reagents for ROH to RX Thionyl chloride SOCl2 + ROH RCl + HCl + SO2 Phosphorus tribromide PBr3 + 3ROH 3RBr + H3PO3 Examples CH3CH(CH2)5CH3 SOCl2 K2CO3 CH3CH(CH2)5CH3 Cl OH (81%) (pyridine often used instead of K2CO3) (CH3)2CHCH2OH PBr3 (CH3)2CHCH2Br (55-60%) Preparation of Alkenes: Elimination Reactions b-Elimination Reactions Overview dehydrogenation of alkanes: X=Y=H dehydration of alcohols: X = H; Y = OH dehydrohalogenation of alkyl halides: X = H; Y = Br, etc. X b C C a Y C C + X Y Dehydrogenation limited to industrial syntheses of ethylene, propene, 1,3-butadiene, and styrene important economically, but rarely used in laboratory-scale syntheses CH3CH3 CH3CH2CH3 750°C 750°C H2C CH2 + H2 H2C CHCH3 + H2 Dehydration of Alcohols Dehydration of Alcohols CH3CH2OH OH H2SO4 160°C H2C CH2 + H2O + H2O H2SO4 140°C (79-87%) CH3 H3C C CH3 OH H2SO4 H3C C heat H3C CH2 (82%) + H2O R' Relative Reactivity R C OH tertiary: most reactive R" R' R C OH H H R C H OH primary: least reactive Regioselectivity in Alcohol Dehydration: The Zaitsev Rule Regioselectivity H2SO4 HO + 80°C 10 % 90 % A reaction that can proceed in more than one direction, but in which one direction predominates, is said to be regioselective. Regioselectivity CH3 CH3 CH3 H3PO4 + OH heat 16 % 84 % A reaction that can proceed in more than one direction, but in which one direction predominates, is said to be regioselective. The Zaitsev Rule When elimination can occur in more than one direction, the principal alkene is the one formed by loss of H from the b carbon having the fewest hydrogens. R R OH C C H CH3 CH2R three protons on this b carbon The Zaitsev Rule When elimination can occur in more than one direction, the principal alkene is the one formed by loss of H from the b carbon having the fewest hydrogens. R R OH C C H CH3 CH2R two protons on this b carbon The Zaitsev Rule When elimination can occur in more than one direction, the principal alkene is the one formed by loss of H from the b carbon having the fewest hydrogens. R R OH C C H CH3 CH2R only one proton on this b carbon The Zaitsev Rule When elimination can occur in more than one direction, the principal alkene is the one formed by loss of H from the b carbon having the fewest hydrogens. R R OH C C H CH3 R CH2R C CH2R R only one proton on this b carbon C CH3 Stereoselectivity in Alcohol Dehydration Stereoselectivity H2SO4 + heat OH (25%) (75%) A stereoselective reaction is one in which a single starting material can yield two or more stereoisomeric products, but gives one of them in greater amounts than any other. The Mechanism of Acid-Catalyzed Dehydration of Alcohols A connecting point... The dehydration of alcohols and the reaction of alcohols with hydrogen halides share the following common features: 1) Both reactions are promoted by acids 2) The relative reactivity decreases in the order tertiary > secondary > primary These similarities suggest that carbocations are intermediates in the acid-catalyzed dehydration of alcohols, just as they are in the reaction of alcohols with hydrogen halides. Dehydration of tert-Butyl Alcohol CH3 H3C C CH3 H3C OH H2SO4 C heat CH2 + H3C first two steps of mechanism are identical to those for the reaction of tert-butyl alcohol with hydrogen halides H2O Mechanism Step 1: Proton transfer to tert-butyl alcohol H (CH3)3C .. O: + H O + .. H H fast, bimolecular H + (CH3)3C O : H + H tert-Butyloxonium ion :O: H Mechanism Step 2: Dissociation of tert-butyloxonium ion to carbocation H + (CH3)3C O : H slow, unimolecular H + + :O: tert-Butyl cation H (CH3)3C Mechanism Step 3: Deprotonation of tert-butyl cation. H H3C +C H + :O: H CH2 H3C fast, bimolecular H H3C C H3C CH2 + H + O: H Carbocations are intermediates in the acid-catalyzed dehydration of tertiary and secondary alcohols carbocations can: react with nucleophiles lose a b-proton to form an alkene Dehydration of Primary Alcohols H2SO4 CH3CH2OH 160°C H2C CH2 + H2O avoids carbocation because primary carbocations are too unstable oxonium ion loses water and a proton in a bimolecular step Mechanism Step 1: Proton transfer from acid to ethanol H .. CH3CH2 O : + H O .. H H fast, bimolecular H + CH3CH2 O : H Ethyloxonium ion H + :O: H Mechanism Step 2: Oxonium ion loses both a proton and a water molecule in the same step. H H + : O : + H CH2 CH2 O : H H slow, bimolecular H + :O H H H + H2C CH2 + :O: H Rearrangements in Alcohol Dehydration Sometimes the alkene product does not have the same carbon skeleton as the starting alcohol. Example OH H3PO4, heat + 3% + 64% 33% Rearrangement involves alkyl group migration CH3 CH3 C CHCH3 + carbocation can lose a proton as shown CH3 3% or it can undergo a methyl migration CH3 group migrates with its pair of electrons to Rearrangement involves alkyl group migration CH3 CH3 CH3 C CHCH3 + 97% CH3 + C CHCH3 CH3 CH3 3% tertiary carbocation; more stable Rearrangement involves alkyl group migration CH3 CH3 CH3 C CHCH3 + 97% CH3 + C CH3 CH3 3% CHCH3 Another rearrangement CH3CH2CH2CH2OH H3PO4, heat CH3CH2CH 12% CH2 + CH3CH CHCH3 mixture of cis (32%) and trans-2-butene (56%) Rearrangement involves hydride shift CH3CH2CH2CH2 CH3CH2CH H + O: CH2 oxonium ion can lose H water and a proton (from C-2) to give 1-butene doesn't give a carbocation directly because primary carbocations are too Rearrangement involves hydride shift CH3CH2CH2CH2 H + O: CH3CH2CHCH3 + H CH3CH2CH CH2 hydrogen migrates with its pair of electrons from C-2 to C-1 as water is lost carbocation formed by hydride shift is Rearrangement involves hydride shift CH3CH2CH2CH2 H + O: CH3CH2CHCH3 + H CH3CH2CH CH2 CH3CH CHCH3 mixture of cis and trans-2-butene Hydride Shift H CH3CH2CHCH2 + O: H H + CH3CH2CHCH2 + H H : O: H Carbocations can... •react with nucleophiles •lose a proton from the b-carbon to form an alkene •rearrange (less stable to more stable) Dehydrohalogenation of Alkyl Halides b-Elimination Reactions Overview dehydrogenation of alkanes: X=Y=H dehydration of alcohols: X = H; Y = OH dehydrohalogenation of alkyl halides: X = H; Y = Br, etc. X b C C a Y C C + X Y b-Elimination Reactions Overview dehydrogenation of alkanes: industrial process; not regioselective dehydration of alcohols: acid-catalyzed dehydrohalogenation of alkyl halides: consumes base X b C C a Y C C + X Y Dehydrohalogenation is a useful method for the preparation of alkenes NaOCH2CH3 Cl ethanol, 55°C (100 %) likewise, NaOCH3 in methanol, or KOH in ethanol Dehydrohalogenation When the alkyl halide is primary, potassium tert-butoxide in dimethyl sulfoxide is the base/solvent system that is normally used. CH3(CH2)15CH2CH2Cl KOC(CH3)3 dimethyl sulfoxide CH3(CH2)15CH (86%) CH2 Regioselectivity KOCH2CH3 Br + ethanol, 70°C 29 % 71 % follows Zaitsev's rule more highly substituted double bond predominates Stereoselectivity KOCH2CH3 ethanol Br + (23%) (77%) more stable configuration of double bond predominates Mechanism of the Dehydrohalogenation of Alkyl Halides: The E2 Mechanism Facts (1) Dehydrohalogenation of alkyl halides exhibits second-order kinetics first order in alkyl halide first order in base rate = k[alkyl halide][base] implies that rate-determining step involves both base and alkyl halide; i.e., it is bimolecular Facts (2) Rate of elimination depends on halogen weaker C—X bond; faster rate rate: RI > RBr > RCl > RF implies that carbon-halogen bond breaks in the rate-determining step The E2 Mechanism concerted (one-step) bimolecular process single transition state –C—H bond breaks component of double bond forms –C—X bond breaks The E2 Mechanism R .. – O .. : H C C : X: .. Reactants The E2 Mechanism R .. – O .. : H C C : X: .. Reactants The E2 Mechanism R d– .. O .. H Transition state C C d– : X: .. The E2 Mechanism R .. O .. H C C .. – : X: .. Products Anti Elimination in E2 Reactions Stereoelectronic Effects Stereoelectronic effect Br KOC(CH3)3 (CH3)3COH (CH3)3C cis-1-Bromo-4-tertbutylcyclohexane (CH3)3C Stereoelectronic effect (CH3)3C trans-1-Bromo-4-tertbutylcyclohexane Br (CH3)3C KOC(CH3)3 (CH3)3COH Stereoelectronic effect cis Br KOC(CH3)3 (CH3)3COH (CH3)3C Rate constant for dehydrohalogenation of cis is 500 times greater than that of trans (CH3)3C Br (CH3)3C trans KOC(CH3)3 (CH3)3COH Stereoelectronic effect cis Br KOC(CH3)3 (CH3)3COH (CH3)3C H H (CH3)3C H that is removed by base must be anti periplanar to Br Two anti periplanar H atoms in cis stereoisomer Stereoelectronic effect trans H Br H (CH3)3C KOC(CH3)3 (CH3)3COH H H (CH3)3C H that is removed by base must be anti periplanar to Br No anti periplanar H atoms in trans stereoisomer; all vicinal H atoms are gauche to Br cis more reactive trans less reactive Stereoelectronic effect An effect on reactivity that has its origin in the spatial arrangement of orbitals or bonds is called a stereoelectronic effect. The preference for an anti periplanar arrangement of H and Br in the transition state for E2 dehydrohalogenation is an example of a stereoelectronic effect. A Different Mechanism for Alkyl Halide Elimination: The E1 Mechanism Example CH3 CH3 CH2CH3 C Br Ethanol, heat H3C CH3 H2C + C CH2CH3 (25%) H C C CH3 H3C (75%) The E1 Mechanism 1. Alkyl halides can undergo elimination in absence of base. 2. Carbocation is intermediate 3. Rate-determining step is unimolecular ionization of alkyl halide. CH3 Step 1 CH3 CH2CH3 C : Br: .. slow, unimolecular CH3 C CH3 + CH2CH3 .. – : Br : .. CH3 Step 2 CH3 C + CH2CH3 – H+ CH3 CH2 + C CH3 CH2CH3 C CH3 CHCH3







































































































































































![Zuccheri [chimica]](http://s1.studyres.com/store/data/004545645_1-6bd8c4a2d3b0941e429f1520d29dd13d-150x150.png)


