Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Quantitative comparative linguistics wikipedia , lookup
Frameshift mutation wikipedia , lookup
Dominance (genetics) wikipedia , lookup
Hardy–Weinberg principle wikipedia , lookup
Computational phylogenetics wikipedia , lookup
Viral phylodynamics wikipedia , lookup
Point mutation wikipedia , lookup
Microevolution wikipedia , lookup
Coalescent Module- Faro July 26th-28th 04
www.coalescent.dk
Monday
H:
The Basic Coalescent
W:
Forest Fire
W:
The Coalescent + History, Geography & Selection
H:
The Coalescent with Recombination
Tuesday
H:
Recombination cont.
W:
The Coalescent & Combinatorics
HW: Computer Session
H:
The Coalescent & Human Evolution
Wednesday
H:
The Coalescent & Statistics
HW: Linkage Disequilibrium Mapping
Zooming in!
(from Harding + Sanger)
3*109 bp
*5.000
(chromosome 11)
b-globin
Exon 1 Exon 2
5’ flanking
6*104 bp
*20
Exon 3
3’ flanking
ATTGCCATGTCGATAATTGGACTATTTTTTTTTT
3*103 bp
*103
30 bp
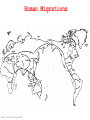
Human Migrations
From Cavalli-Sforza,2001
Data: b-globin from sampled humans.
From Griffiths, 2001
Assume:
1. At most 1
substitution per
position.
2.No recombination
Reducing nucleotide
columns to bipartitions gives a
bijection between data
& unrooted gene trees.
C
G
Simplified model of human sequence evolution.
Past
Rate of common ancestry: 1
Africa
Mutation rate: 2.5
Wait to common ancestry: 2Ne
Present
0.2
Non-Africa
From Griffiths, 2001
Models and their benefits.
Models + Data
1.
probability of data (statistics...)
2.
probability of individual histories
3.
hypothesis testing
4.
parameter estimation
Coalescent Theory in Biology
www. coalescent.dk
Fixed Parameters: Population Structure, Mutation, Selection,
Recombination,...
Reproductive Structure
Genealogies of non-sequenced
data
Genealogies of sequenced data
TGTTGT
Parameter Estimation
Model Testing
CGTTAT
CATAGT
Wright-Fisher Model of Population Reproduction
Haploid Model
i. Individuals are made by
sampling with replacement in the
previous generation.
ii. The probability that 2 alleles
have same ancestor in previous
generation is 1/2N
Assumptions
1. Constant
population size
2. No geography
Diploid Model
3. No Selection
4. No recombination
Individuals are made by
sampling a chromosome
from the female and one
from the male previous
generation with
replacement
10 Alleles’ Ancestry for 15 generations
Waiting for most recent common ancestor - MRCA
Distribution until 2 alleles had a common ancestor, X2?:
P(X2 > 1) = (2N-1)/2N = 1-(1/2N)
1
1
2N
P(X2 = j) = (1-(1/2N))j-1 (1/2N)
P(X2 > j) = (1-(1/2N))j
j
j
2
2
1
1
1
2N
1
2N
Mean, E(X2) = 2N.
Ex.: 2N = 20.000, Generation time 30 years, E(X2) = 600000 years.
P(k):=P{k alleles had k distinct parents}
1
1
2N
Ancestor choices:
k -> any
(2N)k
k -> k
2N *(2N-1) *..* (2N-(k-1))
=:
(2N)[k]
k -> k-1
k -> j
k
(2N)[k-1]
2
Sk, j (2N)[ j ]
Sk,j - the number of ways to group k labelled objects into j groups.(Stirling
Numbers of second kind.
k
For k << 2N:
- / 2N
k
2N[k ]
2
2
P(k)
(k
2N)
1/2N
e
(2N) k
2
Geometric/Exponential Distributions
The Geometric Distribution: {1,..} Geo(p):
P{Z=j)=pj(1-p)
P{Z>j)=pj
E(Z)=1/p.
The Exponential Distribution: R+
Exp (a)
Density: f(t) = ae-at,
P(X>t)= e-at
Properties: X ~ Exp(a)
i.
Y ~ Exp(b) independent
P(X>t2|X>t1) = P(X>t2-t1)
(t2 > t1)
ii.
E(X) = 1/a.
iii.
P(Z>t)=(≈)P(X>t) small a (p=e-a).
iv.
P(X < Y) = a/(a + b).
v.
min(X,Y)
~ Exp (a + b).
Discrete Continuous Time
tc:=td/2Ne
6
6/2Ne
0
k
k
X k is exp[ ] distributed. E(X k ) 1/
2
2
1.0 corresponds to 2N generations
1.0
2N
0
1
4
2
6
5
3
0.0
Adding Mutations
m
mutation pr. nucleotide pr.generation.
L: seq. length
µ = m*L Mutation pr. allele pr.generation. 2Ne - allele number.
Q := 4N*µ -- Mutation intensity in scaled process.
Continuous time
Continuous sequence
Discrete time
Discrete sequence
1/L
sequence
sequence
mutation
Q/2
mutation
Q/2
time
time
1/(2Ne)
coalescence
1
Probability for two genes being
identical:
P(Coalescence < Mutation) = 1/(1+Q).
Note: Mutation rate and population size usually appear together
as a product, making separate estimation difficult.
The Standard Coalescent
Two independent Processes
Continuous: Exponential Waiting Times
Discrete: Choosing Pairs to Coalesce.
Waiting
{1,2,3,4,5}
Coalescing
(1,2)--(3,(4,5))
Exp2
{1,2}{3,4,5}
2
Exp3
{1}{2}{3,4,5}
2
Exp4
{1}{2}{3}{4,5}
2
Exp5
{1}{2}{3}{4}{5}
1
2
3
4
5
2
1--2
3--(4,5)
4--5
Expected Height and Total Branch Length
Time Epoch
Branch Lengths
1
1
2
1/3
1
2
3
k
k
2
1 /
2 k (k - 1)
Expected Total height of tree:
2/(k-1)
Hk= 2(1-1/k)
i.Infinitely many alleles finds 1 allele in finite time.
ii. In takes less than twice as long for k alleles to find 1
ancestors as it does for 2 alleles.
Expected Total branch length in tree, Lk:
2*(1 + 1/2 + 1/3 +..+ 1/(k-1)) ca= 2*ln(k-1)
Kingman
(Stoch.Proc. & Appl. 13.235-248 + 2 other articles,1982)
A. Stochastic Processes on Equivalence Relations.
D ={(i,i);i= 1,..n}
1
if s
Q ={(i,j);i,j=1,..n}
<
t
qs,t =
0
otherwise
This defines a process, Rt , going from to through equivalence relations
on {1,..,n}.
B. The Paint Box & exchangable distributions on Partitions.
C. All coalescents are restrictions of “The Coalescent” – a
process with entrance boundary infinity.
D. Robustness of “The Coalescent”: If offspring distribution is
exchangeable and Var(n1) --> s2 & E(n1m) < Mm for all m, then
genealogies follows ”The Coalescent” in distribution.
E. A series of combinatorial results.
Effective Populations Size, Ne.
In an idealised Wright-Fisher model:
i. loss of variation per generation is 1-1/(2N).
ii. Waiting time for random alleles to find a common
ancestor is 2N.
Factors that influences Ne:
i. Variance in offspring. WF: 1. If variance is higher,
then effective population size is smaller.
ii. Population size variation - example k cycle:
N1, N2,..,Nk.
iii. Two sexes
k/Ne= 1/N1+..+ 1/Nk.
N1 = 10 N2= 1000 => Ne= 50.5
Ne = 4NfNm/(Nf+Nm)I.e. Nf- 10 Nm -1000
Ne - 40
6 Realisations with 25 leaves
Observations:
Variation great close to root.
Trees are unbalanced.
Sampling more sequences
The probability that the ancestor of the sample of size n is in a sub-sample of size k is
(n 1)(k - 1)
(n -1)(k 1)
Letting n go to infinity gives (k-1)/(k+1), i.e. even for quite small samples it is quite large.
Three Models of Alleles and Mutations.
Infinite Allele
Infinite Site
Finite Site
acgtgctt
acgtgcgt
acctgcat
tcctgcat
tcctgcat
Q
Q
Q
acgtgctt
acgtgcgt
acctgcat
tcctggct
tcctgcat
i. Only identity,
non-identity is
determinable
ii. A mutation
creates a new type.
represented by a line.
i. Allele is
represented by a
sequence.
ii. A mutation
always hits a new
position.
ii. A mutation changes
nucleotide at chosen
position.
i. Allele is
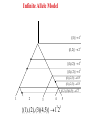
Infinite Allele Model
{(1)} 11
{(1,2)} 21
{(1), (2)} 12
{(1), (2)} 12
{(1), (2,3)} 1121
{(1), (2,3)} 1121
{(1,2), (3)( 4,5)} 1122
1
2
3
4
5
{(1), (2), (3)( 4,5)} 1 2
3 1
Infinite Site Model
Final Aligned Data Set:
0
1
1
1
2
4
3
5
4
5
5
5
6
3
7
2
8
1
0
Number of paths:
1
1
1
2
4
3
2
4
3
5
7
6
7
8
2
4
7
2
6
4
8
14
22
28
2
10
32
50
82
5
2
5
2
5
3
2
1
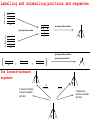
Labelling and unlabelling:positions and sequences
1
2
3
4
5
Ignoring mutation position
Ignoring sequence label
1
2
3
5
4
Ignoring mutation position
{
,
,
Ignoring sequence label
}
The forward-backward
argument
2
5(4 )
4 classes of mutation
events incompatible
with data
1
(4 )
9 coalescence
events incompatible
with data
Infinite Site Model: An example
Theta=2.12
2
3
2
5
3
4
5
9
10
5
14
19
33
Impossible
Ancestral
States
Finite Site Model
Final Aligned Data Set:
acgtgctt
acgtgcgt
acctgcat
tcctgcat
tcctgcat
s s
s
Simplifying assumptions
1) Only substitutions.
s1
s2
TCGGTA
TGGT-T
s1
s2
TCGGA
TGGTT
2) Processes in different positions of the molecule are independent.
3) A nucleotide follows a continuous time Markov Chain.
4) Time reversibility: I.e. πi Pi,j(t) = πj Pj,i(t), where πi is the stationary distribution of i.
This implies that
P( a ) * P
(l )*Pa,N 2 (l2 ) P(N1 )PN1,N 2 (l1 l2 )
a,N1 1
a
a
l1
N1
l2
=
N1
l2+l1
N2
N2
5) The rate matrix, Q, for the continuous time Markov Chain is the same at all times.
Evolutionary Substitution Process
A
t1
e
t2
C
C
Pi,j(t) = probability of going from i to j in time t.
lim e -0
Pi , j (e )
e
qij
lim e -0
Pi ,i (e ) - 1
e
-qii
Jukes-Cantor 69:
TO
FROM
A
A -3*
C
G
T
Total Symmetry.
C
G
-3*
T
-3*
-3*
A. Stationary Distribution: (.25,.25,.25,.25)
B.
Expected number of substitutions: 3t
t
0
P ,t (C, G ) 1 (1 - e-4t )
4
ATTGTGTATATAT….CAG
ATTGCGTATCTAT….CCG
Chimp
Mouse
Fish
Higher Cells
E.coli
History of Coalescent Approach to Data Analysis
1930-40s: Genealogical arguments well known to Wright &
Fisher.
1964: Crow & Kimura: Infinite Allele Model
1968: Motoo Kimura proposes neutral explanation of molecular
evolution & population variation. So does King & Jukes
1971: Kimura & Otha proposes infinite sites model.
1972: Ewens’ Formula: Probability of data under infinite
allele model.
1975: Watterson makes explicit use of
1982:
“The Coalescent”
Kingman introduces “The Coalescent”.
1983: Hudson introduces “The Coalescent with Recombination”
1983: Kreitman publishes first major population sequences.
History of Coalescent Approach to Data Analysis
1987-95: Griffiths, Ethier & Tavare calculates site data
probability under infinite site model.
1994-: Griffiths-Tavaré + Kuhner-Yamoto-Felsenstein introduces
highly computer intensitive simulation techniquees to estimate
parameters in population models.
1996- Krone-Neuhauser introduces selection in Coalescent
1998- Donnelly, Stephens, Fearnhead et al.: Major
accelerations in coalescent based data analysis.
2000-: Several groups combines Coalescent Theory & Gene
Mapping.
2002: HapMap project is started.
Basic Coalescent Summary
i. Genealogical approach to population genetics.
ii. ”The Coalescent” - generic probability distribution on
allele trees.
iii. Combining ”The Coalescent” with Allele/Mutation
Models allows the calculation the probability of data.