Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
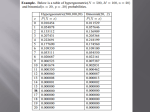
Statistics Behind Differential Gene Expression Arkadipta Bakshi University of Tennessee-Knoxville RNA Sequencing Workshop 26th May, 2016 Overview of the Talk Differential Expression Fold Change Distributions DEseq2 Applications of High-Throughput Sequencing Reading Applications: The sequence itself: Re-sequencing Target-enriched sequencing De-novo assembly Counting Applications: The ability to count the amounts of reads and compare these counts: ChIP Sequencing RNA Sequencing Question: What do we mean by differential expression of a gene? Basis of Differential Expression Differential expression is the assessment of differences in read counts of genes between two or more experimental conditions. Genes are differentially expressed if this difference is statistically significant. Example: There are two samples from the same patient. One sample is from a kidney tumor biopsy. The other sample is a biopsy from the patient's other kidney, which seems to be perfectly healthy tissue. Theoretically, we would expect that the two samples will have different amounts of certain messenger RNA transcripts. It would be interesting to see which transcripts from the tumor are being synthesized at a significantly higher or lower number in the tumor tissue compared to that in the healthy tissue. Why is Differential Expression in RNAseq different from Microarray and other High Throughput Data? Differences in gene expression in microarray data are based on numerical intensity values. Quantitative Metabolome analysis is based on area of the peak generated by each metabolite in the sample. RNAseq is based on sequence read count distributions. RNAseq provides richer information i.e. increased specificity and sensitivity for enhanced detection of differential gene expression. Overview of the Talk Differential Expression Fold Change Distributions DEseq2 Why don’t we just calculate fold change directly? Calculating fold change directly can be misleading. Low counts can appear to have high fold changes while large counts are less sensitive. Question: What are the different methods that have been used to assess differential expression in RNAseq data? Overview of the Talk Differential Expression Fold Change Distributions Modeling read counts with Poisson distribution Overdispersion and the negative binomial distribution DEseq2 Question: Why it might be appropriate to model read counts as a Poisson based-process? Normalization • Comparing genes to each other brings in many more biases – If we are comparing the same gene across two data sets (not two genes to each other), we can make the assumption that length and other biases largely cancel out – Thus we can ignore these issues: • Issue 1: Gene length – At similar expression levels, a longer gene will collect more reads than a shorter gene. • Issue 2: Uniqueness of mapped reads – If one gene has a region that is not unique, many reads are lost. – When compared to another gene of the same length that is entirely unique, no reads are lost. • Issue 3: GC content – If one gene has a much higher GC content or a region of particularly high GC content, the sequencer will produce fewer or no reads from that region. – When compared to another gene of normal GC content, where no reads are lost. Normalization – More units of expression • Raw counts are sometimes altered in other ways to reveal the proportion of transcripts in the original pool of RNA • FPKM = Fragments Per Kilobase of exon per Million Mapped reads (paired end reads) • RPKM = Reads Per Kilobase of exon per million Mapped reads • Used by cufflinks (single end reads) count * 109 transcript length * total reads sequenced Lior Patcher, Models for transcript quantification from RNA-Seq, ArXiv http://arxiv.org/abs/1104.3889 Justification of Poisson Distribution for RNAseq http://www.mi.fu-berlin.de/wiki/pub/ABI/GenomicsLecture13Materials/rnaseq2.pdf Why do we use the poisson distribution vs. the binomial distribution? The binomial distribution is valid when there is a fixed number of events "n" each with a constant probability of success “p". Justification of Poisson Distribution (PD) for RNAseq PD expresses the probability of a given number of events occurring in a fixed interval of time and/or space, if these events occur with a known average rate and independently of the time since the last event . However for poisson distribution, we don’t know the number of "n" trials that will happen. we don’t know how many times success did not happen. we only know the average number of successes per interval. Poisson Distribution (mean = variance) P (x ; µ) = (e − µ )(µx ) X! where x is the number of success and µ is a given region Poisson Distribution • The Poisson model assumes that the mean equals the variance. • Initially confirmed by an RNASeq study with the same initial source of RNA split into multiple lanes of an Illumina GA sequencer (Marioni et al. 2008). – Technical replicates only! • Genuine biological replicates will exhibit higher levels of variation. • Analyzing biologically replicated data with the Poisson model will likely be prone to high false-positive rates due to the underestimation of the true variability (Anders and Huber 2010; Langmead et al. 2010; Robinson and Smyth 2008). Technical Variation = Fits Poisson Mean–variance plot for Marioni et al. dataset (Marioni et al. 2008). The variability in technically replicated RNA-seq data can be adequately captured using a Poisson model. The grey points in this plot shows the mean and pooled variance for each gene, scaled to account for differences in library size between samples. The black line displays the theoretical variance under the Poisson model where the variance is equal to the mean. The red crosses show binned variance, where genes are grouped by mean level. Biological Variation ≠ Fit Poisson Mean–variance plot for the Parikh et al. Dictyostelium dataset (Parikh et al. 2010). The variability in this biologically replicated RNAseq dataset exhibits prominent extraPoisson variability. Restrictions with Poisson Distribution Overdispersion Many studies have shown that the variance grows faster than the mean in RNAseq data. Mean count vs variance of RNA seq data Orange: the fitted observed curve. Purple: the variance implied by the Poisson distribution. Question: How can we address the overdispersion problem during handling of RNAseq data? Negative Binomial Distribution Can be used as a better substitute for an overdispersed poisson. if we define a "1" as failure, all non-"1"s as successes, and we throw a dice repeatedly until the third time “1” appears (r = three failures), then the probability distribution of the number of non-“1”s that had appeared will be a negative binomial. Allows mean and variance to be different. Requires: p – probability of a single success r – the total number of successes Question: What are the different packages that can be used to analyze RNA sequencing data? What to do? • Most people now use the Negative Binomial distribution – Cuffdiff2 <- The only one that deals with DE isoforms – Limma – DESeq2 – EdgeR – SAMSeq Why do we need a distribution? • Why not just use a non-parametric method? – More difficult to show significance with a non-parametric method with few replicates • Rank order statistics will begin working well with ~ 10 biological replicates – SamSeq (http://www.insider.org/packages/cran/samr/docs/SAMseq) Overview of the Talk Differential Expression Fold Change Distributions DEseq2 Modeling dispersion • Now we have a distribution that allows the dispersion to be different from the mean. • But we often still have very low sample numbers (n = 2, 3, 4), which is not good for modeling variance. • A variety of ways to handle this – usually share information across genes to measure variance. • Both DESeq2 and EdgeR assume that genes of similar average expression strength have similar dispersion. – Use this information in slightly different ways to predict reasonable dispersions DEseq2 Accepts raw counts of sequencing reads Requires an associated design formulae Null hypothesis: the expression change in a gene is 0 Calculates differential expression using negative binomial distribution Steps Performed by DEseq Function Estimation of size factors Estimation of dispersion Negative Binomial Generalized Linear Model fitting for βi and Wald statistics. • Generalized linear model is fit for each gene – Flexible - allows for complex designs Plot and fit a curve, adjusting the dispersion parameter toward the curve (shrinking). Likelihood Ratio Test vs. Wald Test Likelihood Ratio Test Compares the likelihood of the data assuming no differential expression (null model) against the likelihood of the data assuming differential expression (alternative model). Estimates two models and compares the fit of one model to the fit of the other. Wald Test: Default Test Uses likelihood ratio but it only estimates one model. Tests the null hypothesis that a set of parameters is equal to some value. Cook’s Distance – Method to Determine the Influential Points Used to remove outliers Measures how much a single sample is influencing the fitted coefficients for a gene. p-values and adjusted p-values for genes deemed as outliers are set to NA in DEseq2 if there are 3 or more replicates. POINTS TO BE NOTED: Points for which Cook's distance is higher than 1 are to be considered as influential. A threshold of 4/N or 4/(N−k−1), where N is the number of observations and k is the number of explanatory variables is also considered. The observations 7 and 16 could be considered as influential. The observation 29 is not substantially different from a couple of other observations. Why do we need to adjust p values? Correct for type 1 error (i.e. false discovery rate) in p values. Adjusted p values Benjamini-Hochberg Needs to adjust for multiple testing (of many genes) Controls false discovery rate Ranks p-values from smallest to largest Assumptions and potential problems: Individual tests are independent of each other May report false negatives What else can DESeq2 do? • Vignette and manual available from Bioconductor site • http://bioconductor.org/packages/release/bioc/html/DESeq2.html • Other Applications: – MA plot: Mean (normalized) expression vs log fold change – Count data transformations: Operates on raw read counts – Heatmap Analysis – Sample clustering: Good for Quality Control (QC). – Principal Components Plot: Use only for QC. Summary RNAseq provides with a richer information, whereas microarray provides only probe specific information. Never calculate fold change directly for RNAseq Data. DEseq2 uses the negative binomial distribution but other distributions can be used. CuffDiff2 seems to work worse than others. Possibly because it has extra statistics to deal with isoforms. (If you would like to deal with DE between isoforms this is probably your best bet). Limma, DESeq(2) and EdgeR are pretty similar (even though limma doesn’t use negative binomial). Biological replicates are better than more depth. When dealing with large datasets it is very important to adjust the p-values to avoid type 1 errors.