Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Gel electrophoresis of nucleic acids wikipedia , lookup
Comparative genomic hybridization wikipedia , lookup
Molecular cloning wikipedia , lookup
Nucleic acid analogue wikipedia , lookup
Cre-Lox recombination wikipedia , lookup
Endogenous retrovirus wikipedia , lookup
Personalized medicine wikipedia , lookup
Deoxyribozyme wikipedia , lookup
Community fingerprinting wikipedia , lookup
Non-coding DNA wikipedia , lookup
Molecular evolution wikipedia , lookup
DNA sequencing wikipedia , lookup
Genome evolution wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
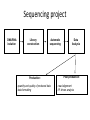
New sequencing technologies in cancer research Alla L Lapidus, Ph.D. Associate Professor Fox Chase Cancer Center Content • Sequencing technologies • Their use in Cancer research • What is available at FCCC • Data quality “REVOLUTIONARY GENOME SEQUENCING TECHNOLOGIES THE $1000 GENOME” (Department of Health and Human Services (DHHS)) 2004 - develop novel technologies that will enable extremely low-cost, high quality DNA sequencing 2009 - the cost to sequence an entire individual human genome to be $1,000 by the end of 2009 and the time required for sequencing less than one week Evolution of sequencing J. Craig Venter, Nature 464, 676-677 (1 April 2010) Sanger sequencing approach vs next-generation (next-gen) approaches Advances: Cheaper (!) no cloning involved => no cloning bias deeper coverage can be obtained Challenges: shorter reads => problems for de-novo assemblies higher error rate (homopolymer related errors in case of 454 and/or IonTorrent) GC-bias still remains slow turn around old bioinformatics tools can not be used and more computer memory and storage space is needed 3-d generation: Price continuing to go down Error rate is lower (Illumina, SOLID) Read lengths increased Bioinformatics is improved but space remains an issue (Amazon cloud etc) • DNA fragmentation • Fragment end repair and end modification (adaptor/linker ligation) • Library enrichment • Sequence DNA amplification NA At Fox Chase Illumina Dense lawn of primers DNA fragment primers Duplicate reads 10x Coverage (depth) Read coverage Reference Clone coverage Cost of the project = f (Turnaround + Read coverage) Read coverage = f (Signal to noise ratio) We need high enough coverage to eliminate errors and better quality reads for lower project cost! Ion Torrent (LifeTechnologies) – 100bp reads, cheap, simple PacBio – read length 1kb The Personal Genome Machine has been designed The PacBio RS system conducts, monitors and for research purposes, offering to charge an iPod analyzes single molecule, realtime (SMRT™) whilst it analyses DNA. It has an iPod dock on top… sequencing reactions. The instrument features high performance optics, automated liquid handling, and The rig uses parallel semi-conductor sensors to an environmental control center, all accessed measure the hydrogen ions produced during DNA through an intuitive touchscreen user interface. replication in real-time (pH…). It is the first machine to use this type of semiconductor technology, while still using traditional integrated fluids and micromachining to translate the information in our DNA into digital information that can be easily measured. So far there are no details as to the price of the device http://www.iontorrent.com/?s_kwcid=TC-12648-4977112303-p-652132722 http://www.pacificbiosciences.com/products/pacbio-rs-system Nanopores for DNA sequencing An attractive strategy for single-molecule DNA sequencing is to pass single-stranded DNA through a nanopore in a graphene monolayer. Here, the rings of carbon atoms in the graphene are depicted as hexagons, and the diameter of the nanopore is about 1.5 nm, corresponding to about 35 hexagonal units. The strand is moving from top to bottom in an applied electric potential, and each of the four DNA bases is shown in a different colour. The DNA could be sequenced by observing the flow of ions through the pore (vertical yellow shading) and recording the distinctive fluctuations of ionic current caused by each type of DNA base as it blocks the ionic flow. Alternatively, fluctuations in a transverse tunneling current (horizontal yellow shading) carried through the graphene, and modulated by DNA passing through the pore, could be measured; the crocodile clips represent electrical connections. One possible problem is that single-stranded DNA can adhere to graphene, as shown. Three scientific papers now report that fluctuations of ionic current can be measured when DNA passes through a graphene nanopore, although the resolution of the measurements is currently insufficient to detect and identify individual bases. http://www.nature.com/nature/journal/v467/n7312/images_article/467164a-f1.2.jpg Summary: - New sequencing technologies allow to sequence genomes faster, at a lower cost and with significantly higher coverage - They change VERY rapidly - Different technologies produce data with different error rates and error profiles -Read length is between 30bp and 500bp -Run time is between 4 hours and 2 weeks -As a result you end up with a tremendous amount of data that needs to be sorted through and analyzed Content • Sequencing technologies • Their use in Cancer research • What is available at FCCC • Data qaulity Sequencing project DNA/RNA isolation Library construction Automatic sequencing Production: - quantity and quality of produced data - data formatting Data Analysis Post-production: - read alignment - PI driven analysis Cancer is a disease of genome alterations. Which alterations can be detected: M. Meyerson, S.Gabriel, G.Getz, Nature Reviews Genetics 11, 685-696 Applications ►Whole genome re-sequencing ►Targeted sequencing (regions, genes, exomes) ►de novo sequencing ►Whole transcriptome sequencing ►miRNA discovery and profiling ►DNA Methylation ►Histone Modification ►DNA-protein interaction Somatic mutations Modified from: M. Meyerson, S.Gabriel, G.Getz, Nature Reviews Genetics 11, 685-696 Recent publications - 1 , Recent publications - 2 Recent publications - 3 The International Cancer Genome Consortium (ICGC) Complexities of cancer genomics 1. Cancer samples differ from the peripheral blood samples that are used for germline genome analysis in their: quantity – for example, diagnostic biopsies from patients contain only few cells quality - DNA/RNA from cancer are often of lower quality (formalin-fixed and paraffin-embedded => increased background mutation rate) purity - mix of cancer and normal genomes 2. Cancers themselves may be highly heterogeneous and composed of different clones that have different genomes 3. “Cancer genomes are enormously diverse and complex. They vary substantially in their sequence and structure compared to normal genomes and among themselves. To paraphrase Leo Tolstoy's famous first line from Anna Karenina: normal human genomes are all alike, but every cancer genome is abnormal in its own way.” – M.Meyerson, S.Gabriel , G.Getz, Nature Reviews Genetics 11, 685-696 (October 2010) 4. To identify somatic alterations in cancer, comparison with matched normal DNA from the same individual is essential. 5. Costly whole-genome sequencing (tumor/normal) is the best approach to discover the full range of genomic alterations — including nucleotide substitutions, structural rearrangements, and copy number alterations — using just this single approach. Cost of computing vs sequencing cost Moore’s law: computers double in power roughly every two years— an increase of more than 30 times over the course of a decade, with reductions in cost. (Moore's law describes a long-term trend in the history of computing hardware. The number of transistors that can be placed inexpensively on an integrated circuit has doubled approximately every two years. The trend has continued for more than half a century and is not expected to stop until 2015 or later.) A map of human genome variation from population-scale sequencing The 1000 Genomes Project Consortium* 2 8 O C T O B E R 2 0 1 0 | VO L 4 6 7 | N AT U R E | 1 0 6 1 Production group People in different aspects of data analysis - wgs of 179 individuals from 4 populations - 2 mother-father-child trio (high coverage) - Exone-targeted sequencing of 697 individuals Content • Sequencing technologies • Their use in Cancer research • What is available at FCCC • Data quality Whole Exome sequencing (WES) at FCCC 1. Exone capture for all or selected genes 2. Exone/exome sequencing 3. Production data QC (data quality, exome coverage) 4. Analysis - Read alignment - Total exome SNPs and small Indels identification (detection and annotation) - dbSNP/1000Genomes filter - synonymous changes filter - additional filters based on the scientific task - manual inspection (with some limitations) - list of candidate genes for PI’s validation Content • Sequencing technologies • Their use in Cancer research • What is available at FCCC • Data quality Homozygous SNPs and indel Poor alignment Missed SNP? Haplotype sequencing If someone has two disease-linked mutations within a single gene, it's difficult to determine with current genome sequencing methods if there is one genetic mistake on the maternal copy and one on the paternal copy or if both variations lie within the same copy of the gene. In the former case, the person has two defective genes, which are likely to cause health problems. In the latter, the person has one good copy of the gene and one bad copy. In many cases, having the good copy can compensate for the defective one. "You lose a lot of information if you look at things at a genotype level versus a haplotype level." Nicholas Schork, Scripps Research Institute, Nature Biotechnology. Ways to haplotype whole genomes 1. University of Washington - combined next-gen sequencing with large insert cloning (fosmids) to achieve a sequenced genome with haplotype information 2. Stanford University - used microfluidics technology in combination with genotyping to obtain haplotype information at the single-cell level 1. Combination of old and new sequencing approaches -made fosmid library from DNA from a HapMap individual (female) of Indian descent - split the library into more than 100 different pools - Barcoded pools - shotgun-sequenced the libraries on the Illumina Genome Analyzer to a mean depth of 2.4-fold per haploid clone. -whole-genome resequencing to search for variants.(Illumina HiSeq 2000 with 50 base paired-end reads, 15-fold coverage. -assembled data into haplotype blocks of different length (>37kb) - phased variants (Genomic phase, the assignment of alleles to homologous chromosomes) J. O. Kitzman Nature Biotechnology (2010) What was detected: (a) Homozygous deletion (top), hemizygous deletion (middle) and inversion (bottom) with fosmid clone support. Deletion calls were made using read depth and paired-read discordance. Inversions were called by paired-read discordance. SNPs within hemizygous deletions appear as stretches of hemizygosity by whole-genome shotgun sequencing. Purple connections indicate the additional support of strand discordance of read pairs spanning genomic DNA and the vector backbone. (b) Novel contigs not present in the reference assembly (red) but detected among clone pool–derived reads (light blue, purple, yellow) are anchored by searching for positions in the reference common to those pools but missing from most or all other pools. This approach anchors 1,733 recently reported insertion sequences including contig GU268019. 2. Single-Cell Approach Whole-genome molecular haplotyping of single cells - use microfluidic device to captured single cells - protease digestion to release the chromosomes - randomly separated the chromosomes into 48 regions - chromosomes were then individually amplified and analyzed with PCR, so that two pools with differing homologous chromosomes could be created - each pool containing one haploid genome, was genotyped using an Illumina SNP array and by creating haplotype blocks the size of a full chromosome. H.C. Fan, J.Wang, A.Potanina, S.R.Quake, Nature Biotechnology, 2010 THANK YOU!