Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
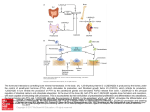
Published OnlineFirst October 26, 2011; DOI: 10.1158/1078-0432.CCR-10-2507 Clinical Cancer Research Molecular Pathways Molecular Pathways: Osteoclast-Dependent and Osteoclast-Independent Roles of the RANKL/RANK/OPG Pathway in Tumorigenesis and Metastasis William C. Dougall Abstract Receptor activator of nuclear factor-kappa B ligand (RANKL) is a TNF ligand superfamily member that is essential for the formation, activation, and function of osteoclasts. RANKL functions via its cognate receptor RANK, and it is inhibited by the soluble decoy receptor osteoprotegerin (OPG). In skeletal metastases, the ratio of RANKL to OPG is upregulated, which leads to increased osteoclast-mediated bone destruction. These changes in the bone microenvironment not only compromise the structural integrity of bone, leading to severe clinical morbidities, but have also been implicated in establishment of de novo bone metastasis and the progression of existing skeletal tumors. Evaluation of RANKL inhibitors, including the fully human antiRANKL antibody denosumab, in patients with cancer has shown reductions in tumor-induced bone resorption activity and successful management of skeletal complications of bone metastases. RANKL also functions as a major paracrine effector of the mitogenic action of progesterone in mouse mammary epithelium, and it has a role in ovarian hormone-dependent expansion and regenerative potential of mammary stem cells. RANKL inhibition attenuates mammary tumorigenesis and pulmonary metastases in mouse models. These data suggest that the contribution of progesterone to increased mammary cancer incidence is mediated, at least in part, by RANKL-dependent changes in the mammary epithelium; RANKL also directly promotes distant metastases. In summary, the antitumor and antimetastatic effects of RANKL inhibition can occur by at least 2 distinct mechanisms, one in the bone via osteoclast-dependent effects, and the second via direct effects on the tumor cells of various origins and/or mammary epithelium. Clin Cancer Res; 18(2); 326–35. 2011 AACR. Background Control of normal bone remodeling by the RANK/RANKL pathway The cells that control bone remodeling include the osteoblast, which deposits new bone, and the osteoclast, a specialized cell of hematopoietic origin that resorbs the inorganic and organic matrix of the bone. Osteoblast and osteoclast function are coordinately regulated in normal bone remodeling. Identification of the receptor activator of nuclear factor-kappa B (RANK), RANK ligand (RANKL), and osteoprotegerin (OPG) pathway in the mid-1990s revealed a key molecular axis for osteoclast formation, function, and survival, and it provided crucial insights into the regulation of normal, physiologic bone remodeling. Functional genomics in mice showed that RANKL Author's Affiliation: Department of Hematology and Oncology Research, Amgen Inc., Seattle, Washington Corresponding Author: William C. Dougall, Department of Hematology and Oncology Research, Amgen Inc., 1201 Amgen Ct. West, Seattle, WA 98119. Phone: 206-265-7553; Fax: 206-217-0494; E-mail: [email protected] doi: 10.1158/1078-0432.CCR-10-2507 2011 American Association for Cancer Research. 326 (TNFSF11), a member of the TNF ligand superfamily, and RANK (TNFRSF11a), the cognate TNFR family receptor for RANKL, were essential for osteoclastogenesis in vivo. A functional interaction between RANKL, expressed by bone stromal cells of the osteoblast lineage, and RANK, expressed by osteoclast precursors of hematopoietic myeloid lineage, is necessary for osteoclast differentiation, survival, and activation. Knockout mice lacking either RANK or RANKL develop significant osteopetrosis resulting from a lack of osteoclasts and absence of bone resorption (1, 2). OPG (TNFRSF11b), another member of the TNFR superfamily, can also bind RANKL and functions as a soluble decoy receptor for RANKL. The critical role of OPG in osteoclastogenesis and bone remodeling was first shown by the increased bone mass as a result of reduced osteoclast numbers observed in transgenic mice overexpressing OPG (3) and, conversely, the osteopenia observed in OPG knockout mice (4). Given that the precise and balanced interaction of RANK, RANKL, and OPG is critical for osteoclastogenesis and, therefore, the maintenance of homeostatic bone remodeling and bone mass, it was hypothesized that this pathway may become dysregulated within the bone during disease states and contribute to pathologic bone loss, such as postmenopausal osteoporosis or cancer-induced bone destruction (Fig. 1). Clin Cancer Res; 18(2) January 15, 2012 Downloaded from clincancerres.aacrjournals.org on June 18, 2017. © 2012 American Association for Cancer Research. Published OnlineFirst October 26, 2011; DOI: 10.1158/1078-0432.CCR-10-2507 RANKL in Tumorigenesis and Metastasis A Physiological bone turnover/remodeling (normal) B Tumor-induced pathologic bone turnover/remodeling (high) Tumor 7 Tumor, dormant micro-metastasis Variety of tumor-derived factors 1 Bone stromal cell Bone stromal cell RANKL 3 Osteoclast Osteoblast Bone resorption Bone formation Expanded tumor mass 6 2 Tumor cell expressing RANKL RANKL 5 ↑ RANKL ↓ OPG Osteoclasts Osteoblasts ↑ Bone formation ↑ Bone resorption 4 Bone destruction (spectrum of lesions) Homeostatic maintenance of bone mass Osteoblastic Mixed osteolytic/ osteoblastic Osteolytic © 2011 American Association for Cancer Research Figure 1. Bone-intrinsic functionality of the RANKL pathway in normal and pathologic osteoclastogenesis. A, normal bone turnover and/or remodeling observed in healthy physiologic systems. Bone stromal cells, including cells of the osteoblast lineage, provide a limited amount of RANKL, which leads to osteoclast differentiation, survival, and activation and subsequent bone resorption. Resorption is balanced by osteoblast-dependent new bone formation. If occult micrometastases were to exist, they would remain dormant because of the low level of bone turnover (37). B, tumor-induced high bone turnover and/or remodeling. (1) A variety of tumor-derived factors will cause increases in RANKL and/or decreases in OPG within the bone stroma. (2) Some tumor cells can also directly produce RANKL. (3) The increased RANKL-to-OPG ratio leads to increased osteoclast formation, survival, and activity, which increases the rate of bone remodeling and/or turnover. (4) Pathologic bone remodeling, characterized by increased osteoclast and osteoblast activities, causes a spectrum of bone lesions in patients with bone metastasis, ranging from predominately osteoblastic to predominately osteolytic. Multiple myeloma gives rise to purely lytic bone lesions. (5) As a result of the increased osteoclast function, many changes occur within the reactive bone microenvironment. These changes include increases in local levels of calcium, release of activated growth factors from the bone matrix, and increased production of growth and/or angiogenic factors by the osteoclast. (6) Increased tumor cell growth and survival, including the outgrowth of dormant micrometastases, occur as a result of these bone matrix–and osteoclast-derived factors. (7) Skeletal tumor cells respond to these bone signals with further production of additional proresorptive factors, generating a feed-forward loop known as the "vicious cycle." A unique feature of skeletal metastases and multiple myeloma is the active involvement of the host, particularly osteoclasts, in the pathophysiologic process leading to bone destruction and skeletal tumor growth. Accelerated osteoclast action and subsequent increased bone resorption have been operatively linked to a full spectrum of skeletal complications (e.g., hypercalcemia, pathologic fracture, bone pain, spinal cord compression, surgery or radiation treatment to the bone, etc.) that result from bone metastases and multiple myeloma. Osteoclast involvement is observed in all types of cancer-induced bone destruction, regardless of the lesions’ radiographic appearance as either osteolytic or osteoblastic, and irrespective of the tumor type of origin (5–7). In addition to causing destructive bone loss and associated clinical sequelae, tumor-induced osteoclasts contribute to the establishment, growth, and survival of tumors. www.aacrjournals.org Products of osteoclastic bone resorption, including matrixderived growth factors [e.g., TGF-b, insulin-like growth factor I (IGF-I)] and elevated calcium levels, increase tumor cell proliferation and survival and induce further production of osteolytic and osteoblastic factors, thereby creating a positive feedback loop known as the "vicious cycle" (7). As osteoclasts are highly related to macrophages, it is perhaps not surprising that many protumorigenic factors can be produced by osteoclasts directly, including growth factors [platelet-derived growth factor a, interleukin 1 (IL-1), TNF, IL-6, IGF-I, BAFF, APRIL] or angiogenic factors [VEGFa, VEGFc, basic fibroblast growth factor (bFGF)], in addition to the production of matrix metalloproteinases (e.g., MMP-7, MMP-9, MMP-14; refs. 8, 9). Thus, the osteoclast may indirectly provide factors that contribute to skeletal tumor establishment and progression, as a consequence of bone matrix turnover, in addition to cellular production, Clin Cancer Res; 18(2) January 15, 2012 Downloaded from clincancerres.aacrjournals.org on June 18, 2017. © 2012 American Association for Cancer Research. 327 Published OnlineFirst October 26, 2011; DOI: 10.1158/1078-0432.CCR-10-2507 Dougall independent of bone resorption. Initial studies of breast cancer by Stephen Paget in 1889 identified the bone as an optimal site (or "soil") for distant metastases ("seed"; ref. 10). It is now clear that many other tumors, including prostate, lung, renal, melanoma, and thyroid, have a high propensity to metastasize to bone, highlighting the skeleton as a fecund metastatic site capable of harboring early tumor colonization and actively promoting the growth of skeletal tumors. The RANKL, RANK, OPG pathway in bone- and cancer-induced bone disease In each of these aforementioned pathologic processes (bone destruction, metastatic establishment, and tumor progression), the tumor has coopted the host by upsetting the balance of 2 key factors that normally govern physiologic osteoclast formation and bone remodeling: RANKL and OPG (Fig. 1). The cancer-induced signals capable of altering the normally well-balanced RANKL-to-OPG ratio can be extremely diverse, reflecting the different sources of RANKL and OPG, the assortment of tumor types that affect bone, and the heterogeneity of individual tumor types (7, 11). More importantly, these diverse signals and mechanisms that elevate osteoclastogenesis and bone destruction converge on the RANKL pathway. Many different cytokines or factors produced by skeletal tumors [e.g., IL-1b, IL-6, IL-8, IL-11, IL-17, macrophage inflammatory protein 1a, TNFa, parathyroid hormone-releasing protein (PTHrP), prostaglandin E (PGE2)] cause increased RANKL production by stromal cells in the bone microenvironment, including cells of the osteoblast lineage. OPG, the normal decoy receptor for RANKL, can also be downregulated by tumors via various mechanisms; reduced synthesis or active degradation of OPG in the bone is observed as a consequence of tumor-derived factors (11). Certain factors in metastatic cancer have dual effects on the RANKL-to-OPG ratio. For instance, PTHrP, IL-1, and PGE2 have been shown to act on the bone stroma and stimulate osteoclast activity by increasing RANKL and decreasing OPG simultaneously (7). T lymphocytes, including activated T cells (12) and CD4þCD25þ T-regulatory cells (T-reg; ref. 13), may be another source of RANKL in bone metastases or other extraskeletal cancer settings in which RANKL may function (see below). Multiple myeloma cells can increase RANKL production in T lymphocytes (14); however, functional evidence for T-cell contribution of RANKL in cancer–bone interactions is lacking, largely because most preclinical bone cancer models are studied in immunocompromised hosts. Increases in RANKL that lead to cancer-induced osteoclastogenesis are not limited to infiltrating immune cells or reactive changes in the bone stroma caused by the tumor but can be contributed by the tumor cells themselves. RANKL expression has also been reported on some tumor types, including breast cancer, prostate cancer, multiple myeloma, and renal carcinoma (as described below). RANKL produced by tumor cells can increase osteoclastogenesis in vitro (15), suggesting that the tumor cell localized in the skeleton 328 Clin Cancer Res; 18(2) January 15, 2012 may also directly contribute an osteoclastogenic signal. It is certainly possible that the bone microenvironment provides signals that can increase RANKL on metastatic tumor cells, which in turn enhances further osteoclast activity and creates a feed-forward loop. For instance, RANKL increases have been observed in prostate tumor cell lines after treatment with TGF-b (16), a product of bone resorption, or upon stimulation of the epithelial–mesenchymal transition (EMT) by TGF-b plus epidermal growth factor treatment or transfection with SNAIL (17). Intriguingly, EMT changes in tumor cells residing in the bone have been linked to a more invasive phenotype, and development of overt bone metastases from dormant micrometastases (17, 18), although a functional role for RANKL in this transition, has not yet been defined. Consistent with the hypothesis that RANKL production by tumor cells accelerates osteoclast formation and bone metastasis is the observation that a high expression level of RANKL in primary renal cell carcinoma is associated with a shorter bone metastasis–free survival (19). A similar relationship between high RANKL expression levels in primary tumors and development of bone metastases has also been described for patients with hepatocellular carcinoma (20). Currently, no definitive data correlate circulating levels of RANKL in serum with bone metastases. This lack of data may reflect the preponderance of the membrane-bound versus soluble form of RANKL, the relatively low levels of RANKL detected in the serum, or the technical limitations of such assays (21). It is also likely that increases in RANKL remain localized to the bone lesion, where focal activation of bone remodeling and increased osteoclast activity are closely juxtaposed to tumor infiltration (22). In contrast, a relationship between the higher ratio of serum RANKL to OPG and a greater extent of bone lesions has been observed in multiple myeloma (23), which may reflect either the extensive tumor cell production of RANKL or the systemic nature of this hematologic tumor. To address the functional contribution of RANKL to cancer-induced bone disease, pharmacologic RANKL inhibitors, such as OPG and RANK-Fc, have been tested in rodent models, and these studies have been the subject of recent reviews (11). Preclinical models of cancer bone metastasis and multiple myeloma typically develop osteolytic, osteoblastic, or mixed lesions after systemic or intratibial injection in rodents. RANKL inhibition has been tested in models representing many different tumor types, including multiple myeloma, breast cancer, prostate cancer, lung cancer, colon cancer, etc., and in each case has been shown to effectively reduce tumor-induced bone lesions (11). Given the specific mechanism of RANKL inhibition, the observed broad activity across lesion types suggests that osteoclastic activity may be a requisite element for both osteolytic and osteoblastic lesions. In addition to the beneficial effect on bone lesions, RANKL blockade has also reduced other tumor-associated sequelae, including bone pain (24) and hypercalcemia of malignancy (25). On the basis of the observed reciprocal feedback (the vicious cycle) occurring between bone and tumor, one Clinical Cancer Research Downloaded from clincancerres.aacrjournals.org on June 18, 2017. © 2012 American Association for Cancer Research. Published OnlineFirst October 26, 2011; DOI: 10.1158/1078-0432.CCR-10-2507 RANKL in Tumorigenesis and Metastasis would predict that reduction of bone resorption would not only reduce the tumor-induced bone destruction but also slow skeletal tumor progression. Studies using preclinical cancer–bone models representing different solid tumors and multiple myeloma have shown a substantial reduction in tumor growth in the bone as a consequence of the potent osteoclast blockade achieved with RANKL inhibition (6, 7, 26). The reductions in skeletal tumor burden upon RANKL inhibition are dependent on the bone microenvironment and are associated with increased tumor cell apoptosis, decreased tumor cell proliferation, and increased survival of tumor-bearing mice (27–30). RANKL inhibition effectively reduces skeletal tumor growth as a monotherapy, but as would be predicted by an approach that targets the bone microenvironment and reduces local growth factor and calcium production, the reduction in skeletal tumor burden is additive when combined with chemotherapy, hormonal therapy, or targeted therapies (28, 31–33). If the "seed and soil" observations and "vicious cycle" hypothesis postulate that the fertile nature of the bone microenvironment actively contributes to the processes of early tumor colonization and metastatic outgrowth, then skeletal metastases might be prevented or delayed through pharmacologic blockade of bone turnover (34), with RANKL inhibition as a potential approach. To test this hypothesis in preclinical models, however, one must consider limitations of experimental bone metastases models, including the observation that skeletal growth of xenografted human tumor cells may ultimately achieve a critical mass capable of self-maintenance independent of the bone microenvironment. To address this limitation, it has been necessary to model tumor–bone interactions at early stages of bone metastasis and colonization using high-sensitivity, small-animal imaging techniques. Prophylactic treatment of mice with the RANKL inhibitor OPG-Fc was employed to reduce baseline osteoclast activity and bone resorption prior to inoculation of breast tumor MDA-231 cells. This strategy subsequently delayed de novo formation of metastatic skeletal tumors as monitored by bioluminescent imaging, presumably by arresting the vicious cycle supporting initial tumor growth (27). The RANKL pathway in normal mammary biology and cancer In addition to severe osteopetrosis, RANK and RANKL knockout mice manifested a lactation defect, revealing an intrinsic functionality of the pathway in mammary epithelium that is also relevant to mammary tumorigenesis and metastases. The failure of RANK or RANKL knockout mice to lactate was due to a marked inability to develop lobuloalveolar mammary structures, which normally undergo a massive expansion under hormonal control during pregnancy and eventually differentiate into milk-secreting tissue. Proliferation and survival of the mammary epithelium are reduced in the absence of a productive RANKL/RANK signal, and transplantation experiments showed this defect to be autonomous to mammary epithelial tissue (35). RANKL expression is greatly increased in luminal mammary www.aacrjournals.org epithelial cells at midgestation pregnancy and can be induced by factors such as prolactin, PTHrP, and progesterone (35). In mice, RANK expression is observed at low levels in luminal and basal cells of the mammary epithelium and becomes more highly expressed at midgestation pregnancy, most selectively at ductal branch points (36). Genetic studies revealed that both RANKL and progesterone receptor (PR) function at similar stages in lactational morphogenesis and that these proteins are colocalized in luminal mammary epithelial cells. Recent experiments have shown that the major mitogenic effect of progesterone occurs via increases in RANKL within PRþ luminal epithelial cells. RANKL, acting as a paracrine factor, then induces mitogenesis of neighboring estrogen receptor (ER)/PR mammary epithelial cells (37–39). Moreover, other recent studies have shown that RANKL can mediate the nonproliferative expansion of the mouse mammary gland via increases in the number and regenerative capacity of mammary stem cells (MaSC). Despite their ER/PR phenotype, MaSC are profoundly responsive to ovarian hormone signaling, and RANKL was identified as a paracrine effector of the progesterone-dependent effects on MaSC occurring during either gestation or estrous cycles (40, 41). RANKL promotes mammary tumorigenesis The fundamental roles of the RANKL pathway in the normal physiology of the mammary gland have significant implications in cancer and tumorigenesis. RANKL-driven hormone (progesterone)-dependent proliferation, survival, and nonproliferative expansion of MaSC could each contribute to mammary cancer initiation, progression, and recurrence (Fig. 2). During normal lactational morphogenesis, it is well characterized that RANK and RANKL adhere to strict, yet overlapping, spatial–temporal expression patterns within the mammary epithelium under control of lactation hormones. However, when RANKL or RANK is overexpressed in the mammary epithelium [via mouse transgenic models using the mouse mammary tumor virus (MMTV) promoter] in the absence of strict hormonal control, inappropriate mammary proliferation is observed (36, 42). Despite the aberrant proliferation (including hyperplasia) induced by an overactive RANKL pathway in these 2 models, spontaneous mammary tumors were not observed in aged virgin mice. However, accelerated preneoplasias and increased mammary tumor formation were observed in MMTV-RANK mice after multiparity or treatment with the carcinogen 7,12-dimethylbenz(a)anthracene (DMBA) in combination with a synthetic progestin hormone [medroxyprogesterone acetate (MPA); ref. 38], settings in which mammary epithelial RANKL expression is increased owing to increased sex hormone levels. Pharmacologic blockade of RANKL decreased incidence and delayed onset of mammary tumors induced by DMBA and MPA in both MMTV-RANK transgenic mice and wild-type mice (38). The reduction in mammary tumor formation was preceded by a reduction in preneoplasias and correlated with rapid and sustained reductions in hormone-induced mammary epithelial proliferation and cyclin D1 expression, as well as Clin Cancer Res; 18(2) January 15, 2012 Downloaded from clincancerres.aacrjournals.org on June 18, 2017. © 2012 American Association for Cancer Research. 329 Published OnlineFirst October 26, 2011; DOI: 10.1158/1078-0432.CCR-10-2507 Potential sources of RANKL Dougall RANKL by hormones (Progesterone, PTHrP, Prolactin) RANKL in preneoplastic or neoplastic cells RANKL in T-cells RANKL-induced effects in tumorigenesis and metastasis RANKL at metastatic sites (lymph node, bone) Bone Mammary stem cell (MaSC) Lung Normal mammary epithelium Preneoplasia Adenocarcinoma Metastasis Mechanism of RANKLdependent effects (in vivo and in vitro) Proliferation Survival Loss of apical/basal polarity Migration/ invasion Anchorageindependent growth Expansion and regenerative capacity of stem cell component © 2011 American Association for Cancer Research Figure 2. Direct protumorigenic and prometastatic activities of RANKL. Using mammary transformation as a model, several different sources of RANKL are indicated, which may impact multiple steps in tumor progression. For instance, hormonal influences (e.g., progesterone, PTHrP, prolactin) may increase RANKL not only in the normal mammary epithelium but also in preneoplastic, tumor, and metastatic lesions. RANKL has also been observed within preneoplastic and neoplastic cells independent of hormone influences. RANKL expressed on T lymphocytes may potentially be important at multiple stages in cancer, from preneoplasias through metastases. Certain metastatic sites, such as the lymph node and bone, are rich sources of RANKL. Different RANKLdependent activities relevant to cancer formation and progression are summarized above. RANKL may function at multiple steps in tumor initiation, progression, and recurrence, including the increased proliferation and survival of normal mammary epithelium and tumor cells, as well as the enhanced numbers and regenerative potential of MaSC or the putative stem cell component of tumors. Other RANKL-dependent activities on tumor cells potentially relevant to distant metastatic spread are reflected by the in vitro observations of enhanced migration and invasion or increased growth in a semisolid medium. increased apoptosis, suggesting that RANKL affects early stages of tumor formation. A similar reduction in mammary tumor formation (also induced by MPA and/or DMBA) was observed by Schramek and colleagues (43) in mice, in which RANK had been selectively deleted from the mammary epithelium by a tissue-specific Cre recombinase– mediated approach. This genetic approach also revealed that the RANKL pathway protects against DNA damageinduced cell death and expands the putative stem cell 330 Clin Cancer Res; 18(2) January 15, 2012 component of mammary tumors. In a model of mammary cancer induced by the c-neu oncogene in the absence of exogenous hormone (MMTV-neu transgenic mice), inhibition of RANKL (beginning at 5 months of age) did not affect median time to spontaneous mammary tumor formation, but it did decrease the number of preneoplastic lesions and mammary tumors (38). Mammary tumor formation was not reduced with another inhibitor of bone resorption [i.e., the nitrogen-containing bisphosphonate zoledronic acid Clinical Cancer Research Downloaded from clincancerres.aacrjournals.org on June 18, 2017. © 2012 American Association for Cancer Research. Published OnlineFirst October 26, 2011; DOI: 10.1158/1078-0432.CCR-10-2507 RANKL in Tumorigenesis and Metastasis (ZA); ref. 38]. In line with the observed reductions in mammary epithelial biomarkers upon RANKL inhibition and the protection against tumors observed in the mammary-selective knockout of RANK, these data confirm direct osteoclast-independent functionality of the RANKL pathway in cancer. A direct role of the RANKL pathway in distant metastases There is also preclinical evidence that blockade of RANKL will reduce distant metastasis, potentially via mechanisms distinct from the bone-intrinsic antiosteoclast effect (Fig. 2). Treatment of transgenic MMTV-neu mice with RANK-Fc significantly decreased spontaneous lung metastases (38). Consistent with this observation, crossing the MMTV-neu mice with RANK heterozygotes reduced the incidence of spontaneous lung metastases (44). Reductions in bone metastases or lung metastases have been achieved with pharmacologic blockade of RANKL in animals bearing RANK-positive melanoma cells (45) or transplanted primary MMTV-neu tumor cells (44). Reciprocally, enhanced lung metastases have been observed upon systemic RANKL exposure in mice bearing orthotopically transplanted tumor cells, originating from either a RANK-positive human breast tumor cell line or tumor cells derived from MMTV-neu mice (44). To gain mechanistic insight into the prometastatic activity of RANKL, considerations of in vitro experiments, in vivo expression patterns of RANK and RANKL, and other potential biologic activities of RANKL are necessary. Expression analyses and treatment of cells in vitro with RANKL have shown functional RANK protein expression on the surface of prostate, breast, osteosarcoma, melanoma, and lung cancer cell lines. In most studies, RANKL does not seem to increase proliferation of RANK-expressing tumor cells (46, 47), although increased cell number and protection against DNA damage-induced cell death, activated by either chemotherapy or g-irradiation, have been observed in certain cell lines (43). In addition, RANKL will affect a variety of other tumor cell behaviors potentially relevant to tumor progression and metastasis, including increased tumor cell growth in semisolid media, loss of apical–basal polarity, and stimulated migration and invasion (38, 43, 45, 46, 48, 49). RANKL treatment has been documented to induce a variety of factors potentially involved in migration, angiogenesis, and invasion, including MMP1, MMP9, the matrix metalloproteinase inducer EMMPRIN/CD47, ICAM-1, IL-6, IL-8, and VEGF (47), and it can decrease expression of the metastasis suppressor serpin 5b/maspin (50). RANKL-dependent promotion of tumor cell migration and invasion (as defined in in vitro experiments described above) may certainly contribute to increased distant metastases observed in vivo. RANKL would be predicted to be highly expressed in normal (or reactive normal) tissues at distant sites of metastases, including peripheral lymph nodes and bone (51), in addition to any RANKL potentially expressed within the primary tumor. In tumor cell lines, RANK expression on tumor cells is not strictly required for www.aacrjournals.org bone metastasis in experimental metastases models. However, 2 in vivo studies are supportive of the skeletal source of RANKL enhancing metastases of RANK-expressing tumors directly: (i) the selective prevention of RANK-expressing tumor cells homing to the bone by RANKL inhibition but not by bisphosphonates (45) and (ii) the increased skeletal growth rate of tumor cells with high RANK expression compared with tumor cell controls with low RANK expression (52). By the same mechanism, RANKL present within lymph nodes or other metastatic sites could then enhance metastatic outgrowth of RANK-positive tumors. Consistent with the above experimental results, a recent analysis reported that high RANK expression in primary breast tumors was associated with lymph node involvement and a higher risk to develop bone metastases (53). Precise mechanisms for the observed reduction in lung metastases achieved with RANKL inhibition may depend upon the model employed. RANKL is not detected within the primary tumor or inflammatory infiltrate in the spontaneous transgenic MMTV-neu mammary tumor model (38), which contrasts with the RANKL expression within tumor-infiltrating T-regs reported after orthotopic transplantation of primary MMTV-neu tumor cells (or a cell line derived from MMTV-neu tumors; ref. 44). Thus, the clear reduction in pulmonary metastases by RANKL inhibition observed in the transgenic MMTV-neu model is not due to RANKL from infiltrating T lymphocytes; it might instead be explained by the overall reduced tumor burden observed in this model (38) and/or other mechanism influencing survival (44) or colonization of metastatic cells. The potential for a direct contribution of RANKL to tumor progression and metastases by tumor-infiltrating T lymphocytes observed in orthotopically transplanted tumors (44) is intriguing, given the earlier observations that RANKL is frequently observed (>65%) in the infiltrating monocytic cells within the stroma of human primary breast tumors (38) and is upregulated in inflammatory, relative to noninflammatory, human breast cancers (54). Interestingly, although there is no evidence (either from genetics or pharmacologic inhibition) that RANKL inhibition is immunosuppressive in vivo (55), RANKL has been shown to promote the activity of T-regs (56) and macrophages (57), cell types that are capable of enhancing tumor progression and metastases. Currently, no studies directly address any function of RANKL on immune components of the tumor microenvironment, but this hypothesis should be considered as a potential protumorigenic and prometastatic mechanism along with the direct effects on tumor cells and/or normal mammary epithelium outlined above. Clinical-Translational Advances Targeting RANKL to inhibit tumor-induced osteoclasts was shown in preclinical proof-of-concept experiments to be a rational approach for the prevention and treatment of skeletal complications of malignancy, including metastatic colonization of the bone. Pharmacodynamic bone resorption biomarkers, including the N-telopeptide of type I Clin Cancer Res; 18(2) January 15, 2012 Downloaded from clincancerres.aacrjournals.org on June 18, 2017. © 2012 American Association for Cancer Research. 331 Published OnlineFirst October 26, 2011; DOI: 10.1158/1078-0432.CCR-10-2507 Dougall collagen (NTX), are useful in the translational evaluation of an osteoclast inhibitor. NTX is a product of bone degradation and a marker of elevated bone resorption, which is measured as the ratio of urinary NTX to creatinine (uNTX/ Cr). Elevated levels of uNTX/Cr have been associated with increased risk for experiencing skeletal complications, disease progression, and death in patients with bone metastases (58). Early versions of RANKL antagonists included recombinant forms of OPG [e.g., Fc-OPG or OPG-Fc (AMGN 0007)]. The first demonstration of biologic activity of RANKL inhibition in patients with cancer was observed with patients with multiple myeloma and breast carcinoma who had radiographic evidence of lytic or mixed bone disease. OPG-Fc produced rapid dose-related declines in bone resorption biomarkers (including uNTX/Cr levels; ref. 59). However, clinical development of OPG forms by Amgen was terminated because of the relatively short halflife of OPG-Fc in patients with cancer and questions about the potential safety risk of a neutralizing immune response against endogenous OPG. Another variant of OPG (CEP37251; Cephalon) was also being developed; however, the phase I clinical study in healthy postmenopausal women was apparently terminated (60). A RANKL antibody form derived from camelidae (i.e., llamas) termed ALX-0141 (Ablynx) has been tested in a phase I study of healthy postmenopausal women (61). Amgen has developed a fully human antibody against RANKL (denosumab, AMG 162), which has shown greater selectivity to human RANKL, favorable pharmacokinetics, and ease of manufacturing compared with OPG molecules. Denosumab is a fully human, immunoglobulin G2 (IgG2) monoclonal antibody, which binds human RANKL with high affinity (KD ¼ 3 pmol/L) and, as a fully human protein, would not be predicted to engender an immune response in patients (62). It binds to soluble and membrane-bound human RANKL and nonhuman primate RANKL, but not mouse RANKL. It does not cross-react with other TNF ligand family members. Similar to OPG, denosumab functions as a reversible RANKL antagonist, preventing RANKL interaction with RANK and inhibiting osteoclast differentiation, activation, and survival. Denosumab is administered via a subcutaneous route. A phase I evaluation in patients with multiple myeloma or breast cancer metastatic to bone showed a pharmacodynamic effect of denosumab within 24 hours after a single subcutaneous dose (59). Compared with a single intravenous dose of the antiresorptive bisphosphonate, pamidronate 90 mg, the decrease in bone turnover markers (including uNTX/Cr levels) produced by denosumab was similar in magnitude but more sustained. In these patients, denosumab was generally well tolerated with no serious adverse events or antidenosumab antibodies detected. Phase II studies evaluated the safety and efficacy of different dosing regimens of denosumab in patients with cancer, and they informed dose and schedule selection for subsequent phase III trials. Also, phase II and phase III studies established the unique mode of action of denosu- 332 Clin Cancer Res; 18(2) January 15, 2012 mab relative to other bone-targeted drugs, specifically nitrogen-containing bisphosphonates (nBP), such as pamidronate and ZA. Bisphosphonates bind to hydroxyapatite bone mineral surfaces, where they inhibit mature osteoclasts locally, at sites of bone resorption (63). Intravenous nBPs have been effective in prevention of skeletal complications in cancer, but safety and tolerability issues exist, including renal toxicity and acute-phase reactions. High levels of uNTX/Cr persist in a substantial proportion of patients with breast and prostate cancers and other solid tumors while on intravenous nBP treatment. As a result, these patients may experience skeletal-related events (including pathologic fracture, radiation, or surgery to bone, or spinal cord compression; ref. 58), indicating the need for improved therapies. Denosumab reduced bone turnover markers in patients with cancer in 2 phase II studies. One study in women with bone metastases from breast cancer concluded that more patients treated with denosumab had bone turnover suppression compared with intravenous nBP (64). In a second randomized phase II study, denosumab was tested in patients with bone metastasis from prostate, breast, or other solid tumors, or multiple myeloma, who had persistent elevated levels of uNTx/Cr levels, despite being treated with intravenous nBP (65). Of these patients, 71% who received denosumab had reduced levels of uNTx/Cr compared with 29% of patients who continued to receive intravenous nBP. Furthermore, suppression of serum TRAP5b, an osteoclast marker, was 2.5-fold greater in patients treated with denosumab compared with patients continuing treatment with intravenous nBP. In both studies, the safety profile of denosumab was consistent with a cancer population receiving systemic antineoplastic treatment. Evaluation of the phase II results, using a population pharmacokinetic–pharmacodynamic model, suggested that a dose of 120 mg denosumab given every 4 weeks would suppress uNTX/Cr levels by more than 90% in most patients (64), and pharmacokinetic analyses of serum denosumab levels indicated a mean half-life of approximately 30 days (66). Altogether, these phase II studies clearly defined the efficacy of denosumab in patients with cancer, independent of the tumor type, and established the ability of denosumab to control hyperactive bone resorption. Skeletal-related events are serious irreversible complications of tumors that metastasize to the bone. To evaluate the ability of denosumab to prevent skeletal-related events, 3 large phase III clinical studies were done in patients who had bone metastases from breast cancer (67), castrationresistant prostate cancer (CRPC; ref. 68), and any other advanced cancer (excluding breast and prostate) or multiple myeloma (69). Each trial was a randomized, double-blind, double-dummy, active controlled comparison of s.c. denosumab 120 mg with i.v. ZA 4 mg (adjusted for creatinine clearance) every 4 weeks. The primary endpoint for each study was time to first on-study skeletal-related event. In the breast cancer trial (n ¼ 2,046), denosumab significantly delayed the time to first skeletal-related event by 18% versus Clinical Cancer Research Downloaded from clincancerres.aacrjournals.org on June 18, 2017. © 2012 American Association for Cancer Research. Published OnlineFirst October 26, 2011; DOI: 10.1158/1078-0432.CCR-10-2507 RANKL in Tumorigenesis and Metastasis ZA (P ¼ 0.01, superiority) and reduced the risk of multiple skeletal-related events by 23% versus ZA (P ¼ 0.001, superiority), showing the durability of the positive effect on skeletal complications (67). In men with CRPC and bone metastases (n ¼ 1,904), denosumab significantly delayed the time to first on-study skeletal-related event by 18% compared with ZA (P ¼ 0.008, superiority) and the time to first and subsequent skeletal-related event (P ¼ 0.008, superiority; ref. 68). Finally, Henry and colleagues (69) reported that denosumab delayed time to first on-study skeletal-related event (HR 0.84; P ¼ 0.0007, noninferiority) compared with ZA in patients with bone metastases from a variety of solid tumors (excluding prostate and breast cancer, but representing more than 50 different tumor types) or multiple myeloma. For all 3 studies, time to disease progression and overall survival rates were similar between the denosumab- and ZA-treated cohorts. The overall rate of onstudy adverse events was also similar between the 2 cohorts, including infrequent observations of osteonecrosis of the jaw typically associated with previously reported risk factors. Rates of adverse events potentially associated with renal toxicity and acute-phase reactions were elevated in patients treated with ZA, whereas a greater incidence of hypocalcemia was observed in the denosumab cohorts. Hypocalcemia was manageable, was not associated with significant clinical sequelae, and was consistent with denosumab’s mechanism of action. These 3 identically designed pivotal clinical studies were the basis upon which denosumab achieved approval by the U.S. Food and Drug Administration in November 2010 for prevention of skeletalrelated events in patients with bone metastases from solid tumors. Conclusions The early findings of an essential role of RANKL in physiologic osteoclastogenesis was the fundamental rationale for targeting this pathway as a treatment for skeletal complications in cancer, and a therapeutic antibody, denosumab, has been successfully developed in the clinic for this application in patients with solid tumor bone metastases. As preclinical experiments have shown, the interference with the bone microenvironment and potent reduction in bone resorption with a RANKL inhibitor also has potential utility in the inhibition of early metastatic colonization. Consistent with this hypothesis, denosumab was recently shown to be effective in delaying development of bone metastasis in men with CRPC (70), and this study represents the first large randomized study to show that targeting the bone microenvironment has this effect in patients with cancer. This hypothesis is also currently being addressed in women with early-stage breast cancer at high risk of disease recurrence to determine whether denosumab in combination with standardof-care adjuvant and/or neoadjuvant cancer treatment will improve bone metastasis–free and disease-free survival compared with standard-of-care treatment alone (71). www.aacrjournals.org Although more recent preclinical basic research in rodents has indicated that RANKL can promote a spectrum of effects relevant to cancer initiation, progression, and metastasis via direct effects on either tumor cells or the mammary epithelium, significant gaps remain in the translation of these more recent findings to the clinic. The prominent role of RANKL as a paracrine effector of progesterone action in the mouse mammary epithelium clearly has implications in humans, given the potential role for progesterone specifically as a risk factor in human breast cancer, as supported by its well-established mitogenic effects in the breast (72) and by large epidemiologic studies (73). However, fundamental differences exist in the anatomy and hormone responsiveness of the rodent mammary gland compared with the primate, which may limit the utility of the preclinical findings. Data linking the RANKL pathway (RANKL, RANK, and OPG) expression with hormone exposure and breast density and proliferation in humans, or in a suitable nonhuman primate model, are needed to show relevance of the progesterone and RANKL pathway association. To address any association of the RANK/RANKL pathway with cancer risk or progression, genome-wide association studies or analysis of mRNA or protein expression may be helpful, but each approach has its limitations. Using a candidate gene approach, a recent genetic association study has associated a single nucleotide polymorphism in the RANK gene with breast cancer risk (74). In addition to progesterone, multiple other potential stimuli of RANKL in cancer may exist (e.g., PTHrP, prolactin, tumor stromal cells, infiltrating T lymphocytes), and further studies are necessary not only to verify the many diverse sources of RANKL with well-validated methodologies but also to determine whether any of these RANKL sources are associated with disease outcome. Likewise, it will be crucial to define any tumor subpopulations that express RANK, again using well-validated and specific approaches, and to relate RANK expression with additional disease outcomes. Recent analysis of RANK mRNA expression in human breast cancer biopsies indicates relatively increased expression in the basal tumor subtype and an association of higher RANK mRNA expression with poor survival (53). Finally, the identification of tumor-specific (or mammary epithelial-specific) biomarkers of a RANKL response would aid in the translational evaluation of this drug in extraskeletal tissues and, ultimately, the testing of the current preclinical hypotheses in patients. Disclosure of Potential Conflicts of Interest W. C. Dougall: employment and stockholder, Amgen Inc. Acknowledgments I would like to acknowledge the editorial assistance of Albert Rhee and Geoff Smith. I would also like to thank Michelle Blake, Dan Branstetter, Allison Jacob, and Lanny Kirsch for their critique of this article. Received August 16, 2011; revised October 5, 2011; accepted October 6, 2011; published OnlineFirst October 26, 2011. Clin Cancer Res; 18(2) January 15, 2012 Downloaded from clincancerres.aacrjournals.org on June 18, 2017. © 2012 American Association for Cancer Research. 333 Published OnlineFirst October 26, 2011; DOI: 10.1158/1078-0432.CCR-10-2507 Dougall References 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. 17. 18. 19. 20. 334 n-Cardo Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordo C, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003;3:537–49. Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T, et al. RANK is essential for osteoclast and lymph node development. Genes Dev 1999;13:2412–24. €thy R, Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Lu et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell 1997;89:309–19. Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, et al. osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev 1998;12:1260–8. Yonou H, Ochiai A, Goya M, Kanomata N, Hokama S, Morozumi M, et al. Intraosseous growth of human prostate cancer in implanted adult human bone: relationship of prostate cancer cells to osteoclasts in osteoblastic metastatic lesions. Prostate 2004;58:406–13. Roodman GD. Mechanisms of bone metastasis. Discov Med 2004;4: 144–8. Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer 2002;2:584–93. Cappellen D, Luong-Nguyen NH, Bongiovanni S, Grenet O, Wanke C, Susa M. Transcriptional program of mouse osteoclast differentiation governed by the macrophage colony-stimulating factor and the ligand for the receptor activator of NFkappa B. J Biol Chem 2002;277:21971–82. Zhang Q, Guo R, Lu Y, Zhao L, Zhou Q, Schwarz EM, et al. VEGF-C, a lymphatic growth factor, is a RANKL target gene in osteoclasts that enhances osteoclastic bone resorption through an autocrine mechanism. J Biol Chem 2008;283:13491–9. Paget S. The distribution of secondary growths in cancer of the breast. Lancet 1889;133:571–3. Roodman GD, Dougall WC. RANK ligand as a therapeutic target for bone metastases and multiple myeloma. Cancer Treat Rev 2008;34: 92–101. Anderson DM, Maraskovsky E, Billingsley WL, Dougall WC, Tometsko ME, Roux ER, et al. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 1997;390: 175–9. Totsuka T, Kanai T, Nemoto Y, Tomita T, Okamoto R, Tsuchiya K, et al. RANK-RANKL signaling pathway is critically involved in the function of CD4þCD25þ regulatory T cells in chronic colitis. J Immunol 2009;182: 6079–87. Giuliani N, Colla S, Sala R, Moroni M, Lazzaretti M, La Monica S, et al. Human myeloma cells stimulate the receptor activator of nuclear factor-kappa B ligand (RANKL) in T lymphocytes: a potential role in multiple myeloma bone disease. Blood 2002;100:4615–21. Zhang YH, Heulsmann A, Tondravi MM, Mukherjee A, Abu-Amer Y. Tumor necrosis factor-alpha (TNF) stimulates RANKL-induced osteoclastogenesis via coupling of TNF type 1 receptor and RANK signaling pathways. J Biol Chem 2001;276:563–8. Zhang J, Lu Y, Dai J, Yao Z, Kitazawa R, Kitazawa S, et al. In vivo realtime imaging of TGF-beta-induced transcriptional activation of the RANK ligand gene promoter in intraosseous prostate cancer. Prostate 2004;59:360–9. Zhau HE, Odero-Marah V, Lue HW, Nomura T, Wang R, Chu G, et al. Epithelial to mesenchymal transition (EMT) in human prostate cancer: lessons learned from ARCaP model. Clin Exp Metastasis 2008;25: 601–10. Buijs JT, Kuijpers CC, van der Pluijm G. Targeted therapy options for treatment of bone metastases; beyond bisphosphonates. Curr Pharm Des 2010;16:3015–27. Mikami S, Katsube K, Oya M, Ishida M, Kosaka T, Mizuno R, et al. Increased RANKL expression is related to tumour migration and metastasis of renal cell carcinomas. J Pathol 2009;218:530–9. Sasaki A, Ishikawa K, Haraguchi N, Inoue H, Ishio T, Shibata K, et al. Receptor activator of nuclear factor-kappaB ligand (RANKL) expression in hepatocellular carcinoma with bone metastasis. Ann Surg Oncol 2007;14:1191–9. Clin Cancer Res; 18(2) January 15, 2012 21. Bowsher RR, Sailstad JM. Insights in the application of research-grade diagnostic kits for biomarker assessments in support of clinical drug development: bioanalysis of circulating concentrations of soluble receptor activator of nuclear factor kappaB ligand. J Pharm Biomed Anal 2008;48:1282–9. 22. Kitazawa S, Kitazawa R. RANK ligand is a prerequisite for cancerassociated osteolytic lesions. J Pathol 2002;198:228–36. 23. Terpos E, Szydlo R, Apperley JF, Hatjiharissi E, Politou M, Meletis J, et al. Soluble receptor activator of nuclear factor kappaB ligandosteoprotegerin ratio predicts survival in multiple myeloma: proposal for a novel prognostic index. Blood 2003;102:1064–9. 24. Honore P, Luger NM, Sabino MA, Schwei MJ, Rogers SD, Mach DB, et al. Osteoprotegerin blocks bone cancer-induced skeletal destruction, skeletal pain and pain-related neurochemical reorganization of the spinal cord. Nat Med 2000;6:521–8. 25. Capparelli C, Kostenuik PJ, Morony S, Starnes C, Weimann B, Van G, et al. Osteoprotegerin prevents and reverses hypercalcemia in a murine model of humoral hypercalcemia of malignancy. Cancer Res 2000;60:783–7. 26. Li X, Liao J, Park SI, Koh AJ, Sadler WD, Pienta KJ, et al. Drugs which inhibit osteoclast function suppress tumor growth through calcium reduction in bone. Bone 2011;48:1354–61. 27. Canon JR, Roudier M, Bryant R, Morony S, Stolina M, Kostenuik PJ, et al. Inhibition of RANKL blocks skeletal tumor progression and improves survival in a mouse model of breast cancer bone metastasis. Clin Exp Metastasis 2008;25:119–29. 28. Miller RE, Roudier M, Jones J, Armstrong A, Canon J, Dougall WC. RANK ligand inhibition plus docetaxel improves survival and reduces tumor burden in a murine model of prostate cancer bone metastasis. Mol Cancer Ther 2008;7:2160–9. 29. Vanderkerken K, De Leenheer E, Shipman C, Asosingh K, Willems A, Van Camp B, et al. Recombinant osteoprotegerin decreases tumor burden and increases survival in a murine model of multiple myeloma. Cancer Res 2003;63:287–9. 30. Zheng Y, Zhou H, Brennan K, Blair JM, Modzelewski JR, Seibel MJ, et al. Inhibition of bone resorption, rather than direct cytotoxicity, mediates the anti-tumour actions of ibandronate and osteoprotegerin in a murine model of breast cancer bone metastasis. Bone 2007;40: 471–8. 31. Canon J, Bryant R, Roudier M, Coxon A, Dougall W. RANKL inhibition plus tamoxifen blocks ERþ breast tumor growth in bone metastases and prevents tumor-induced bone loss. In: Proceedings of the IXth International Meeting on Cancer Induced Bone Disease; 2009 Oct 28– 31; Arlington, VA. 2009. 32. Canon J, Bryant R, Roudier M, Osgood T, Jones J, Miller R, et al. Inhibition of RANKL increases the anti-tumor effect of the EGFR inhibitor panitumumab in a murine model of bone metastasis. Bone 2010;46:1613–9. 33. Holland PM, Miller R, Jones J, Douangpanya H, Piasecki J, Roudier M, et al. Combined therapy with the RANKL inhibitor RANK-Fc and rhApo2L/TRAIL/dulanermin reduces bone lesions and skeletal tumor burden in a model of breast cancer skeletal metastasis. Cancer Biol Ther 2010;9:539–50. € wik CW, Wetterwald A, 34. van der Pluijm G, Que I, Sijmons B, Buijs JT, Lo et al. Interference with the microenvironmental support impairs the de novo formation of bone metastases in vivo. Cancer Res 2005;65: 7682–90. 35. Fata JE, Kong YY, Li J, Sasaki T, Irie-Sasaki J, Moorehead RA, et al. The osteoclast differentiation factor osteoprotegerin-ligand is essential for mammary gland development. Cell 2000;103:41–50. 36. Gonzalez-Suarez E, Branstetter D, Armstrong A, Dinh H, Blumberg H, Dougall WC. RANK overexpression in transgenic mice with mouse mammary tumor virus promoter-controlled RANK increases proliferation and impairs alveolar differentiation in the mammary epithelia and disrupts lumen formation in cultured epithelial acini. Mol Cell Biol 2007;27:1442–54. 37. Beleut M, Rajaram RD, Caikovski M, Ayyanan A, Germano D, Choi Y, et al. Two distinct mechanisms underlie progesterone-induced Clinical Cancer Research Downloaded from clincancerres.aacrjournals.org on June 18, 2017. © 2012 American Association for Cancer Research. Published OnlineFirst October 26, 2011; DOI: 10.1158/1078-0432.CCR-10-2507 RANKL in Tumorigenesis and Metastasis 38. 39. 40. 41. 42. 43. 44. 45. 46. 47. 48. 49. 50. 51. 52. 53. 54. 55. 56. 57. proliferation in the mammary gland. Proc Natl Acad Sci U S A 2010;107:2989–94. Gonzalez-Suarez E, Jacob AP, Jones J, Miller R, Roudier-Meyer MP, Erwert R, et al. RANK ligand mediates progestin-induced mammary epithelial proliferation and carcinogenesis. Nature 2010;468:103–7. Mukherjee A, Soyal SM, Li J, Ying Y, He B, DeMayo FJ, et al. Targeting RANKL to a specific subset of murine mammary epithelial cells induces ordered branching morphogenesis and alveologenesis in the absence of progesterone receptor expression. FASEB J 2010;24:4408–19. Asselin-Labat ML, Vaillant F, Sheridan JM, Pal B, Wu D, Simpson ER, et al. Control of mammary stem cell function by steroid hormone signalling. Nature 2010;465:798–802. Joshi PA, Jackson HW, Beristain AG, Di Grappa MA, Mote PA, Clarke CL, et al. Progesterone induces adult mammary stem cell expansion. Nature 2010;465:803–7. Fernandez-Valdivia R, Mukherjee A, Ying Y, Li J, Paquet M, DeMayo FJ, et al. The RANKL signaling axis is sufficient to elicit ductal sidebranching and alveologenesis in the mammary gland of the virgin mouse. Dev Biol 2009;328:127–39. Schramek D, Leibbrandt A, Sigl V, Kenner L, Pospisilik JA, Lee HJ, et al. Osteoclast differentiation factor RANKL controls development of progestin-driven mammary cancer. Nature 2010;468:98–102. Tan W, Zhang W, Strasner A, Grivennikov S, Cheng JQ, Hoffman RM, et al. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature 2011;470: 548–53. Jones DH, Nakashima T, Sanchez OH, Kozieradzki I, Komarova SV, Sarosi I, et al. Regulation of cancer cell migration and bone metastasis by RANKL. Nature 2006;440:692–6. Armstrong AP, Miller RE, Jones JC, Zhang J, Keller ET, Dougall WC. RANKL acts directly on RANK-expressing prostate tumor cells and mediates migration and expression of tumor metastasis genes. Prostate 2008;68:92–104. Rucci N, Millimaggi D, Mari M, Del Fattore A, Bologna M, Teti A, et al. Receptor activator of NF-kappaB ligand enhances breast cancerinduced osteolytic lesions through upregulation of extracellular matrix metalloproteinase inducer/CD147. Cancer Res 2010;70:6150–60. Chen LM, Kuo CH, Lai TY, Lin YM, Su CC, Hsu HH, et al. RANKL increases migration of human lung cancer cells through intercellular adhesion molecule-1 up-regulation. J Cell Biochem 2011;112:933–41. Sabbota AL, Kim HR, Zhe X, Fridman R, Bonfil RD, Cher ML. Shedding of RANKL by tumor-associated MT1-MMP activates Src-dependent prostate cancer cell migration. Cancer Res 2010;70:5558–66. Luo JL, Tan W, Ricono JM, Korchynskyi O, Zhang M, Gonias SL, et al. Nuclear cytokine-activated IKKalpha controls prostate cancer metastasis by repressing Maspin. Nature 2007;446:690–4. Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 1998;93:165–76. Tometsko M, Jones J, Miller R, Roudier M, Dougall W, Chaisson-Blake M. Efficacy of a RANKL inhibitor, OPG-Fc, relative to zoledronic acid to inhibit bone metastasis of a RANK-expressing human breast cancer cell line. In: Proceedings of the IXth International Meeting on Cancer Induced Bone Disease; 2009 Oct 28–31; Arlington, VA. 2009. Santini D, Schiavon G, Vincenzi B, Gaeta L, Pantano F, Russo A, et al. Receptor activator of NF-kB (RANK) expression in primary tumors associates with bone metastasis occurrence in breast cancer patients. PLoS ONE 2011;6:e19234. Lerebours F, Vacher S, Andrieu C, Espie M, Marty M, Lidereau R, et al. NF-kappa B genes have a major role in inflammatory breast cancer. BMC Cancer 2008;8:41. Ferrari-Lacraz S, Ferrari S. Do RANKL inhibitors (denosumab) affect inflammation and immunity? Osteoporos Int 2011;22:435–46. Loser K, Mehling A, Loeser S, Apelt J, Kuhn A, Grabbe S, et al. Epidermal RANKL controls regulatory T-cell numbers via activation of dendritic cells. Nat Med 2006;12:1372–9. Breuil V, Schmid-Antomarchi H, Schmid-Alliana A, Rezzonico R, Euller-Ziegler L, Rossi B. The receptor activator of nuclear factor (NF)-kappaB ligand (RANKL) is a new chemotactic factor for human monocytes. FASEB J 2003;17:1751–3. www.aacrjournals.org 58. Coleman RE, Major P, Lipton A, Brown JE, Lee KA, Smith M, et al. Predictive value of bone resorption and formation markers in cancer patients with bone metastases receiving the bisphosphonate zoledronic acid. J Clin Oncol 2005;23:4925–35. 59. Body JJ, Greipp P, Coleman RE, Facon T, Geurs F, Fermand JP, et al. A phase I study of AMGN-0007, a recombinant osteoprotegerin construct, in patients with multiple myeloma or breast carcinoma related bone metastases. Cancer 2003;97[Suppl]:887–92. 60. ClinicalTrials.gov. Bethesda (MD): NIH. Single ascending-dose study to characterize the safety, pharmacokinetics, and pharmacodynamics of CEP-37251 in healthy postmenopausal women 2010. Available from: http://clinicaltrials.gov/show/NCT01159873. 61. van de Wetering de Rooij J, Lyssens C, ten Holder S, D'Artois J, Weeke-Klimp A, Ulrichts H, et al. Safety, pharmacokinetics and efficacy of anti-RANKL nanobody ALX-0141 in healthy postmenopausal women. Ann Rheum Dis 2011;70[Suppl 3]:136. 62. Kostenuik PJ, Nguyen HQ, McCabe J, Warmington KS, Kurahara C, Sun N, et al. Denosumab, a fully human monoclonal antibody to RANKL, inhibits bone resorption and increases BMD in knock-in mice that express chimeric (murine/human) RANKL. J Bone Miner Res 2009;24:182–95. 63. Baron R, Ferrari S, Russell RG. Denosumab and bisphosphonates: different mechanisms of action and effects. Bone 2011;48:677–92. 64. Lipton A, Steger GG, Figueroa J, Alvarado C, Solal-Celigny P, Body JJ, et al. Randomized active-controlled phase II study of denosumab efficacy and safety in patients with breast cancer-related bone metastases. J Clin Oncol 2007;25:4431–7. 65. Fizazi K, Lipton A, Mariette X, Body JJ, Rahim Y, Gralow JR, et al. Randomized phase II trial of denosumab in patients with bone metastases from prostate cancer, breast cancer, or other neoplasms after intravenous bisphosphonates. J Clin Oncol 2009;27:1564–71. 66. Body JJ, Facon T, Coleman RE, Lipton A, Geurs F, Fan M, et al. A study of the biological receptor activator of nuclear factor-kappaB ligand inhibitor, denosumab, in patients with multiple myeloma or bone metastases from breast cancer. Clin Cancer Res 2006;12:1221–8. 67. Stopeck AT, Lipton A, Body JJ, Steger GG, Tonkin K, de Boer RH, et al. Denosumab compared with zoledronic acid for the treatment of bone metastases in patients with advanced breast cancer: a randomized, double-blind study. J Clin Oncol 2010;28:5132–9. ~o R, Brown J, Karsh L, et al. 68. Fizazi K, Carducci M, Smith M, Damia Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: a randomised, double-blind study. Lancet 2011;377:813–22. 69. Henry DH, Costa L, Goldwasser F, Hirsh V, Hungria V, Prausova J, et al. Randomized, double-blind study of denosumab versus zoledronic acid in the treatment of bone metastases in patients with advanced cancer (excluding breast and prostate cancer) or multiple myeloma. J Clin Oncol 2011;29:1125–32. 70. Smith M, Saad F, Coleman R, Shore N, Fizazi K, Tombal B, et al. Denosumab to prolong bone metastasis-free survival in men with castrate-resistant prostate cancer: Results of a global phase 3, randomized, double-blind trial. In: Proceedings of the American Urological Association Annual Meeting; 2011 May 14–19; Washington, DC. 2011. 71. Goss PE, Barrios CH, Bell R, Finkelstein D, Iwata H, Martin M, et al. A randomized, double-blind, placebo-controlled multicenter phase III study comparing denosumab with placebo as adjuvant treatment for women with early-stage breast cancer who are at high risk of disease recurrence (D-CARE). J Clin Oncol 2011;29:TPS152. 72. Hofseth LJ, Raafat AM, Osuch JR, Pathak DR, Slomski CA, Haslam SZ. Hormone replacement therapy with estrogen or estrogen plus medroxyprogesterone acetate is associated with increased epithelial proliferation in the normal postmenopausal breast. J Clin Endocrinol Metab 1999;84:4559–65. 73. Chlebowski RT, Hendrix SL, Langer RD, Stefanick ML, Gass M, Lane D, et al. WHI Investigators. Influence of estrogen plus progestin on breast cancer and mammography in healthy postmenopausal women: the Women's Health Initiative Randomized Trial. JAMA 2003;289:3243–53. 74. Bonifaci N, Palafox M, Pellegrini P, Osorio A, Benitez J, Peterlongo P, et al. Evidence for a link between TNFRSF11A and risk of breast cancer. Breast Cancer Res Treat 2011;129:947–54. Clin Cancer Res; 18(2) January 15, 2012 Downloaded from clincancerres.aacrjournals.org on June 18, 2017. © 2012 American Association for Cancer Research. 335 Published OnlineFirst October 26, 2011; DOI: 10.1158/1078-0432.CCR-10-2507 Molecular Pathways: Osteoclast-Dependent and Osteoclast-Independent Roles of the RANKL/RANK/OPG Pathway in Tumorigenesis and Metastasis William C. Dougall Clin Cancer Res 2012;18:326-335. Published OnlineFirst October 26, 2011. Updated version Cited articles Citing articles E-mail alerts Reprints and Subscriptions Permissions Access the most recent version of this article at: doi:10.1158/1078-0432.CCR-10-2507 This article cites 70 articles, 24 of which you can access for free at: http://clincancerres.aacrjournals.org/content/18/2/326.full.html#ref-list-1 This article has been cited by 3 HighWire-hosted articles. Access the articles at: /content/18/2/326.full.html#related-urls Sign up to receive free email-alerts related to this article or journal. To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at [email protected]. To request permission to re-use all or part of this article, contact the AACR Publications Department at [email protected]. Downloaded from clincancerres.aacrjournals.org on June 18, 2017. © 2012 American Association for Cancer Research.