Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
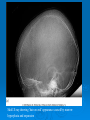
Anemia Anemia Definition. It is an abnormal decrease in the number of circulating RBCs, Hb conc., and hematocrit (PCV). It is not a disease itself but is a symptom of another disorder. It is important to consider the following developmental variations when evaluating an infant or child for anemia: 1-Hb level and PCV are relatively high in the newborn; these values subsequently decline, reaching a nadir at approximately 7 weeks of age for the premature infant and at 2 - 3 ms of age for the term infant. (This condition is referred to as the “physiologic anemia” of infancy or anemia of prematurity) . Total Hb concentration and hematocrit rise gradually during childhood . Normal values in children Age Hb(g/dl) Retic.% cord blood 14-22 5-7 2week 13-20 1 3 month 9.5-14.5 1 6 m- 6yr 7-12 yr 10.5-14.5 11-16 1 1 Adult male 12-16 1.6 Adult female 14-18 1.6 2-Hb F is the major Hb of prenatal and early postnatal life. At cord blood ,Hb F values approached 70% then it decline postnatally; by 9 to 12 months of age, the Hb F values represent <2% of the total Hb concentration. 3-Mean corpuscular volume (MCV) is relatively high during the neonatal period but declines during the latter part of infancy. The MCV is lowest during infancy, gradually increasing with age during childhood, reaching adult levels during adolescence. Classification In clinical practice, anemias are classified according to the morphologic appearance (i.e., color and size) of RBC on peripheral smear as well as the MCV. 1-Hypochromic, microcytic (small, pale RBCs; a low MCV) 2-Macrocytic (large RBCs; a high MCV) 3-Normochromic, normocytic (cells of normal size and shape; a normal MCV) Hypochromic, microcytic anemias Defect. Hypochromic, microcytic RBCs indicate impaired synthesis of the heme or globin components of Hb. Defective heme synthesis may be the result of iron deficiency, lead poisoning, chronic inflammatory disease, pyridoxine deficiency, sideroblastic anemia, or copper deficiency. Defective globin synthesis is characteristic of the thalassemia syndromes. Evaluation. Laboratory studies that are useful in evaluating the hypochromic, microcytic anemia 1- Serum ferritin 2-Total s. iron-binding capacity. 3- Soluble transferrin receptor (sTR). 4- Quantitative measurements of the Hb A1 , Hb A2 and Hb F levels. Normal Hypochromic microcytic Iron deficiency anemia IDA The commonest cause of iron deficiency in children is: 1- Inappropriate diet. 2- Blood loss is uncommon. Iron deficiency occurs from 6 months of age onwards when the child’s total body mass is expanding in the face of an inadequate iron intake causes 1-Nutritional iron deficiency usually develops when rapid growth puts excessive demands on iron stores. This is seen mainly during: A-Infancy, when iron stores at birth are inadequate due to LBW or when the diet is composed exclusively of milk or cereals with low iron content B-Adolescence, when a rapid growth spurt often coincides with a diet of suboptimal iron content (this is a particular problem in girls, who also lose iron with menses) 2-Iron deficiency resulting from blood loss. A-Prenatal iron loss can result from extrusion of fetal blood either into the maternal circulation (fetomaternal transfusion) or into the circulation of a twin (twin-to-twin transfusion). B-Perinatal bleeding may result from obstetric complications such as placental abruption or placenta previa. C-Postnatal blood loss may be of an obvious cause (e.g., after surgery or due to trauma) or may be occult, as occurs in idiopathic pulmonary hemosiderosis, parasitic infestations, polyps, or inflammatory bowel disease. Clinical features. ID is most commonly seen between 6 and 24 months of age. The typical patient is on a diet consisting almost exclusively of milk. Symptoms. Although mild iron deficiency is relatively asymptomatic, as it becomes more severe, the infant manifests 1-irritability 2-anorexia 3-lethargy 4-pica (eating non-food stuffs) 5-apathy 6-easy fatigability. Signs. On physical examination, the milk-fed infant is 1-fat 2-pale 3-other findings include tachycardia and a systolic murmur. If the anemia is very severe, there may be signs of congestive heart failure . 4-other signs (such as koilonychia or angular cheilitis) are very rare. ID causes serial changes in the blood before anemia develops. Serum ferritin is reduced and eventually a microcytic, hypochromic anemia results. Usually the MCV (mean cell volume) and MCH (mean cell hemoglobin) fall before the Hb, but the changes can occur together. The MCHC (mean cell Hb concentration) is less useful . stages of iron depletion Diagnosis 1-CBC: Anemia may vary from very mild to very severe, depending on the degree and duration of ID. Small, pale RBCs are evident on the peripheral smear; the reduction in MCV, MCH and MCHC is usually proportional to the severity of the anemia. 2-The serum iron level is decreased, whereas the iron-binding capacity (transferrin level) is increased, and the percentage of saturation is low (usually <15%). The serum ferritin level is decreased (which is a reflection of low iron stores in the bone marrow), and the sTR level is increased. Differential diagnosis 1.Anemia of chronic disease or ‘anemia of inflammation’ (modification of iron regulation by the inflammatory response). 2.Thalassemia traits , these require quantitation of Hb A2 and F, and not simply Hb electrophoresis. 3.Sideroblastic anemias . 4-Lead poisoning. Iron deficiency and neuropsychological effects ID early in life will affects brain iron content and distribution, leading to neurotransmitter & behavioral alterations. IDA is significantly associated with poorer scores in developmental testing when compared with controls, particularly in coordination and spatial orientation skills. Therapy IDA can be managed by administration of iron. This can be provided by the oral route at a dosage of 6 mg/kg/day of elemental iron for a period of 2 to 3 months after the Hb level has returned to normal; this allows replenishment of tissue iron stores. Dietary counseling must be simultaneously provided to caregivers to give the patient adequate amounts of dietary iron. Dietary iron occurs in two forms , heme and nonheme. Heme iron (in meat, fish and poultry) is well absorbed and its bioavailability is not affected by other dietary factors. Non-heme iron is less well absorbed and its bioavailability is affected by dietary factors because of the way it is bound in foods. It is present in beans, peanut butter, green leafy vegetables, dried fruit and fortified breakfast cereals. Absorption of iron is enhanced by vitamin C and proteins, but is inhibited by a number of constituents of food and drink, for example tannins (in tea and legumes), phytates (in unrefined cereals), phosphates (in eggs), oxalates (in spinach) and polyphenols (in spinach, coffee). Failure to respond to iron therapy, the commonest reason is due to failure of adherence. Although many preparations may be prescribed three times a day, better adherence may be achieved with a single daily dose or twice daily dosing. Types of iron 1. Iron salts (e.g. sulphate, fumarate, gluconate and glycine sulphate). 2. Polysaccharide iron complex. It has major advantages in pediatric practice . They do not stain the teeth and it can be mixed with milk or juice without altering absorption. In general there are fewer GIT side-effects, and they are sugar-free. Perhaps most importantly, the child usually likes them. Anemia of inflammation and chronic disease The anemia of chronic disease is associated with a variety of disorders, including: 1-Chronic inflammatory disease (e.g., Crohn disease , juvenile inflammatory arthritis) 2-Chronic infection (e.g. T.B) 3-Malignancy 4-A mild & transient form of anemia of inflammation may occur following infections, including common viral infections Iron is not released from its storage sites in the macrophages; thus, it is unavailable for Hb synthesis in developing erythroblasts. A modest decrease in the survival of RBCs and a relatively limited erythropoietin response to the anemia also contribute to the development of anemia. Diagnosis The anemia is mild in degree (i.e. Hb is 7–10 g/dL) often with hypochromic,microcytic indices. As in IDA, the serum iron level is reduced. However, in contrast with IDA, the iron-binding capacity is normal or reduced, and the serum ferritin( which is acute-phase reactant) level is increased or normal. Therapy The anemia resolves when the underlying disease process is treated adequately. Therapy with medicinal iron is unnecessary unless concomitant iron deficiency is present. Megaloblastic anemias MA Folate deficiency Etiology MA is very rare indeed in children but when it does occur it is most commonly due to folate deficiency. Unlike iron, folate stores are relatively labile and in constant need of replenishment. Folates are required for nucleic acid synthesis and 1-carbon unit transfer in all cells of the body, particularly growing tissues. MA occurs after 2–3 mo on a folate-free diet. Rapid growth, fever, infection, diarrhea or hemolysis all increase folate requirements and may further deplete the stores to the level of clinical deficiency. Folate is absorbed in the upper jejunum by an active transport mechanism that is impaired in malabsorption states, particularly celiac syndrome. Various drugs are associated with deficiency of folate, e.g. phenytoin, barbiturates, methotrexate, and TMP. Maternal folate deficiency predisposes to neural tube defects and possibly to other congenital abnormalities including Down syndrome. Congenital deficiencies of several enzymes in the folate pathway are described. Clinical features and diagnosis The presentation, like many hematological disorders, is nonspecific. Folate deficiency will produce macrocytosis (which can be masked by associated iron deficiency in conditions with malabsorption); more severe forms will be associated with leucopenia and thrombocytopenia. Hypersegmented neutrophils on the blood film is an important clue. Serum and red cell folate levels should be requested to confirm the Dx Treatment It is straightforward with oral folic acid in a dose of 1–5 mg/d & should be continued for several months. Where demand for folate remains high (e.g. in chronic HA) lifelong supplementation may be required. There is often a dramatic clinical response within a few days with a reticulocytosis by the end of a week. Vitamin B12 deficiency It is very rare indeed in childhood. The infant usually has an insidious onset of pallor, lethargy and anorexia, often with neurological symptoms. With the popularity of vegetarianism, maternal dietary deficiency may produce profound deficiency in infancy with neurological sequelae and is currently the commonest cause of infantile B12 deficiency. It may occur in older children as part of a more generalized GIT disease with malabsorption. B12 deficiency has been reported in infants whose mothers have undergone gastric bypass procedures for obesity, and those whose mothers are in the early stages of traditional pernicious anemia. B12 deficiency occurs in early infancy due to congenital defects in the absorption or metabolic pathway. The diagnosis should be considered in any infant who develops pancytopenia with MA in the first 3 years of life. Diagnosis The blood picture is indistinguishable from folate deficiency – there is often a pancytopenia with macrocytosis. The serum vitamin B12 level will be low. Treatment The usual dose of vitamin B12 (as hydroxocobalamin) for children is 100 μg, given intramuscularly, three times a week until the hemoglobin is normal, followed by 100 μg monthly thereafter. Some disorders may be successfully treated with oral B12 therapy. The neurological defects may take longer to recover. Congenital pure RBC aplasia (Diamond-Blackfan syndrome) a lifelong disorder, usually presents in the first few months of life or at birth with severe anemia and mild macrocytosis or a normocytic anemia. It is due to a deficiency of BM red blood cell precursors. More than a third of patients have short stature. Many pts respond to corticosteroid treatment, but must receive therapy indefinitely. Pts who do not respond to steroid treatment are transfusion dependent and are at risk of the multiple complications of long-term transfusion therapy, especially iron overload. These pts have a higher rate of developing leukemia or other hematologic malignancies than the general population. Transient Erythroblastopenia of Childhood(TEC) a normocytic anemia caused by suppression of RBC synthesis, usually appears after 6 month of age in an otherwise normal infant. Viral infections are thought to be the trigger, although the mechanism leading to RBC aplasia is poorly understood. The onset is gradual, but anemia may become severe. Recovery usually is spontaneous. Differentiation from Diamond-Blackfan syndrome, in which erythroid precursors also are absent or diminished in the BM, may be challenging. Transfusion of packed RBCs may be necessary if the anemia becomes symptomatic before recovery. Thalassemias Definition. Thalassemias are hereditary hemolytic anemias characterized by decreased or absent synthesis of one or more globin subunits of the Hb molecule. α-Thalassemia results from reduced synthesis of α-globin chains. β-thalassemia results from reduced synthesis of βglobin chains. An imbalance in globin chain production is a hazard to the RBC because excess unpaired globin chains produce insoluble tetramers that precipitate, causing membrane damage. This makes RBCs susceptible to destruction within the reticuloendothelial system of the BM (resulting in ineffective erythropoiesis) as well as the RES of the liver and spleen (resulting in hemolytic anemia). The types and quantities of different Hb in infancy & adulthood Type of Hb Notation HbA HbF HbA2 α2β2 α2γ2 α2δ2 Normal % at birth 20 80 1 Normal % in older children 98 1 2 α-Thalassemias α-Thalassemias are usually the result of gene deletion. α-Thalassemia variants are found most often in populations of African or East Asian ancestry. Normally there are four α-globin genes; clinical manifestations of α-thalassemia variants reflect the number of genes affected 1-α-thalassemia major: 4 genes deleted (Hb Bart ) , Hydrops fetalis/death in utero . 2-Hemoglobin H disease:3 genes deleted, (Hb H ), baby born with severe anemia, mild jaundice & splenomegaly 3-α-Thalassemia minor : 2 genes deleted, Mild anemia . 4-Silent carrier :one gene deleted, No anemia β-Thalassemias The clinical phenotype of β-thalassemia is related to the degree of globin chain imbalance. 1-Heterozygous β-thalassemia (β-thalassemia minor). 2-Homozygous β-thalassemia (β-thalassemia major, Cooley anemia, and intermedia). Heterozygous β-thalassemia (β-thalassemia minor) Clinical features. 1- Mild anemia (Hb about 10 gm/dl) 2- Normal growth and development. 3- Blood film: Hypochromia, microcytosis, and anisocytosis. 4- Hb electrophoresis shows elevation of the Hb A2 level and, sometimes, elevation of the Hb F level. Therapy: No treatment is necessary. It is important, however, that thalassemia minor is distinguished from ID to prevent inappropriate therapy with medicinal iron. Folic acid may be given. Genetic counseling is also important. Homozygous β-thalassemia Homozygous β-thalassemia (β-thalassemia major, Cooley anemia, and intermedia). Patients who have this form of anemia are usually of Mediterranean background. Defect. Molecular defects range from complete absence of β-globin synthesis (genotype β0/β0) to partial reduction in the gene product from the affected locus (genotype β+/β+). Clinical features: beginning in the middle of the first year of life 1- the infant manifests a progressively severe HA & jaundice . 2- marked HSM. 3- FTT 4- BM hyperplasia produces characteristic features such as tower skull, frontal bossing, maxillary hypertrophy with prominent cheekbones, and overbite. 5- Hemochromatosis: Even in the untransfused state, iron overload develops in thalassemic pt because of hyperabsorption of dietary iron. The iron load becomes even greater with chronic transfusion therapy. When the BM storage capacity for iron is exceeded, iron accumulates in parenchymal organs such as the liver, heart, pancreas, gonads, and skin, producing the complications of hemochromatosis (“bronzed diabetes”). Many patients succumb to congestive HF , hypoparathyroidism, hypogonadism , DM , liver cirrhosis and short stature. Investigations: 1-CBC : hypochromic microcytic anemia, with nucleated RBC and retics count commonly less than 8% which is inappropraitely low to the degree of anemia due to ineffective rythropoiesis). 2-Elevated unconjucated bilirubin. 3-On Hb electrophoresis, Hb A is either markedly decreased or totally absent. Of the total Hb concentration, 30% to 90% is Hb F. 4-BM hyperplasia is seen in bone XR. 5-Elevated S.ferretin & transferrin saturation. Skull X-ray showing ‘hair on end’ appearance caused by marrow hyperplasia and expansion Treatment: 1- The mainstay of treatment is transfusion with packed RBCs using irradiated CMV –ve blood, a post transfusion Hb level of 9.5-10 gm/dl is the goal. 2- In an effort to prevent hemochromatosis, pts who receive chronic transfusion regimens are treated with chelating agents (e.g., deferoxamine, deferiprone) that promote iron removal from the body through excretion in the urine & stool. Deferoxamine is given subcutaneously over 10-12 hrs,5-6 days a week. Side effects of deferoxamine includes: a-ototoxisity. b-retinal changes. c-bone dysplasia with truncal shortening. d-Yersinia bacteria over growth. 3- Splenectomy is usually considered when transfusion requirements exceed 250 mL/kg/year. 4- bone marrow transplantation can cure the patients . 5- Genetic counseling . HEMOLYTIC ANEMIAS HA Hemolysis : an increased rate of RBC destruction with a shortening of the normal life span of the cell from the normal 120 days to as little as a few days in severe hemolysis. The marrow can increase erythrocyte production 6-8 fold so mild degrees of hemolysis are not associated with anemia. Severe hemolysis can lead to a rapid and profound fall in Hb, and be life threatening. It is suspected when polychromatic cells are seen on the blood film (reticulocytes). It is usually associated with raised blood unconjugated bilirubin and splenomegaly . Hemolysis can be caused by inherited or acquired disorders. Diagnosis of the cause of the hemolysis is made according to the family history; clinical features and red cell morphology that together will indicate what further laboratory tests are required. Marrow examination is generally unnecessary. The child with hemolysis may be pale with fluctuating jaundice (usually mild) and splenomegaly. Pigment gallstones may complicate the disorder ,HA should always be excluded in a child with stones. Aplastic crises may occur, usually precipitated by parvovirus infection which leads to reticulocytopenia and anemia. Parvovirus infection typically produces severe anemia sometimes requiring transfusion, and a modest thrombocytopenia and leucopenia. Hemolytic anemia The major categories of HA are: 1-Immunemediated. 2-Membrane defect (Spherocytosis). 3-Enzyme defect(G6PD def). 4-Hb defect (SCA,Thalassemia). Membrane defects Hereditary spherocytosis (HS) HS It is caused by a defect in the skeleton of the RBC membrane that generally affects the spectrin component. The characteristic finding is increased numbers of spherocytes in the peripheral blood. It is inherited as A.D in 75% of cases, other cases inheritant as A.R or new mutation. Pathology In AD type the defect may be in either beta spectrin,ankyrin or protein 3. In AR form the defect is in either -protein or protein 4-2. This defect affect the RBC skeleton leading to budding of RBC memb, This bud is removed rapidly by the RES leading to loss of RBC surface area.The stretching ability of the RBC is very limited which lead to RBC rupture. The spherocyte have no easy transit through the splenic cord because of its shape this will affect glucose metabolism and decrease pH of RBC. A, polychromatic cell; B, microspherocyte Clinical pictures 1-May be asymptomatic with mild anemia & discovered accidentally. 2-The severe form may be started at early infancy with neonatal jaundice, severe anemia , splenomegaly & chronic blood transfusion. 3-Clinical pictures of complication, which are: A-Bone marrow aplasia after Parvo virus B19 infection. This v. infect erythroid cells in the BM & with arrest in development. The aplasia last for 10-14 days during which time the Hb drops by 50% with reticulocytopenia. B-Hyper hemolysis after some viral infection lead to increasing anemia, reticulocytosis &jaundice. C-Delayed growth and sexual developments. D-Pigmented gallstones. Laboratory diagnosis 1-CBC:A-anemia. B-high MCHC>36. C-high retic. count. D-normal MCH & MCV. E-spherocytes in peripheral blood. 2-Incubated osmotic fragility test is elevated. 3-High indirect bilirubin. 4-Decrease habtoglobin. 5-Membrane protein analysis for difficult cases Management 1-In mild anemia with normal growth and development,we only observe the child and limit the intervention to folic acid tablet 1 mg/day. 2-Splenectomy for any child with chronic anemia or growth failure but we should defer it till the age of 5 yr to minimize the risk of sepsis & we should vaccinate the child by pneumococcal , H. influenza & menengiococcal vaccines at least 2 weeks before splenectomy & give the child prophylactic penicillin after splenectomy until at least 18 yr of age. In some reports,partial splenectomy appears to improve the anemia & maintain splenic function. 3-Elective cholecystectomy for symptomatic gallstones. Glucose-6-phosphate dehydrogenase(G6PD) deficiency G6PD deficiency is the most common RBC metabolic disorder. It is usually transmitted in an X-linked recessive fashion. Defects. The two prototypic forms are: The A- variant is found mainly in the black population and is associated with an isoenzyme that deteriorates rapidly (it has a half-life of 13 days). The Mediterranean variant is found mainly in individuals of Greek and Italian descent and is associated with almost complete absence of enzyme activity, even in young cells, due to extreme instability (it has a half-life of several hours). Pathogenesis G6PD-deficient cells do not generate an amount of reduced glutathione that is sufficient to protect the RBCs from oxidant agents. Exposed sulfhydryl groups of Hb are oxidized, predisposing the molecule to denaturation. The heme and globin moieties dissociate, with the globin precipitating as Heinz bodies. The damaged RBCs are then removed by the RES; severely damaged cells may lyse intravascularly. HA results from oxidative damage to the RBCs as consequence of the loss of the protective effect of the enzyme G6PD. The prevalence of G6PD def. is related to the prevalence of malaria as in Africa, it also high in mediterranean area.The incidence of P falceparum parasite is lower in G6PD def. patients. The half life of the enz in normal RBC is 60 days,the mature RBC cannot synthesize the enz. The younger RBCs are relatively more resistant to hemolysis . The deficient RBC hemolysed when exposed to exogenous factors.The particles of the denaturated Hb,Heinz body,attach to the cell membrane causing irreversible damage and lysis, most of lysis occur intravascularly causing hemoglobinemia & hemoglobinuria. There may be an extravascular hemolysis which explain the splenomegaly in some cases. Favism is the classical cause of acute hemolysis in G6PD def. Fava bean contain the beta glycosides vicine & convicine These substances may undergo auto-oxidation as part of their metabolism, producing free O2 radicals. Acute hemolysis that occurs upon exposure to Fava is characterized by: 1-unpredictable(only 25%of adult at risk develop hemolysis). 2-Influence of dose and body weight. 3-Maturity of the bean. 4-Quality of bean(raw beans more than cooked, frozen or canned beans) 5-The activity of beta glucosidases in the bean & intestinal mucosa. Drug induced hemolysis Many drugs and chemicals have been associated with hemolysis in G6PD def patient,These substances have the ability to stimulate the pentose phosphate PW in RBC which can lead to oxidation of NADPH . Infection-caused hemolysis During the process of phagocytosis of bacteria there is a release of peroxides by the phagocytosing granules, these peroxides lead to release of O radicals. Clinical presentation There are three primary clinical presentation: 1-Neonatal jaundice. 2-Acute hemolysis beyond the neonatal period. 3-Chronic hemolysis (congenital nonspherocytic HA). 1-Neonatal jaundice: Acute hemolysis is characterized by onset of jaundice on the first few days of life that is out of the proportion to the degree of anemia. All infant with G6PD def develop NJ. NJ in G6PD def neonate may be an exaggerated physiological j or may be due to acute hemolysis caused by inciting agent like inf., drugs, naphthalene ball used in stored cloth diapers in developing country. 2-Acute hemolysis: Most pt with G6PD def. are asymptomatic until exposed to inciting agent at which time they may develop hemolysis.The onset of hemolysis is usually within 24-48 hr of exposure. The initial manifestation may include abdominal pain,vomiting or diarrhea,tea colored urine (hemoglobinuria),Jaundice, pallor with symptoms of anemia. Examination usually reveals: anemia , jaundice splenomegaly and hepatomegaly & in severe case symptom of heart failure. Lab.finding in acute hemolysis include: 1-Normochromic normocytic anemia with anisopoikilocytosis, with few spherocyte . 2-Reticulocytosis. 3-Presence of bite cells (RBC bitten by macrophages). 4-Presence of hemigoast cells(RBC with uneven Hb distribution). 5-By using supravital stain we find inclusion bodies called Heinz body(denatured Hb). 6-Low serum haptoglobin. 7-High uncong. bilirubin. 8-Hemoglobinuria . 9- hemoglobinemia. Diagnosis of G6PD def 1-Antenatal diagnosis can be made by performing G6PD assay on amniotic fl cells or from chorionic villi biopsy and DNA assay. 2-Screening test:semiquantitative test,this test should not be done during acute hemolysis because it lead to normal value.The usual cut of deficiency value is <30% of normal activity. 3-Quantitative assay: done several weeks after acute hemolysis when reticulocyte return to normal. 4-G6PD electrophoresis. Differential diagnosis: 1-AIHA. 2-Malaria induced hemolysis. 3-Hepatitis Management of acute severe hemolysis 1-Removal of the inciting agent. 2-Brisk hydration to ensure adequate urine output. 3-blood transfusion. 4-Folic acid tablet 1mg/d for one month. 3-Chronic hemolysis Small minority of pt develop chronic H.Clinically all pts are male,presented with: 1-Neonatal j with anemia. 2-Jaundice and anemia. 3-splenomegaly. 4-Gallstone. 5-Acute hemolysis after exposure to oxidative agent. Lab. Finding: 1-CBC:anemia,reticulocytosis & polychromasia. 2-Low haptoglobin. 3-Hyberbilirubinemia. Management 1-Folic acid administration. 2-Blood transfusion if indicated. Drugs induce hemolysis in G6PD: ANALGESICS/ANTIPYRETICS acetophenetidin , amidopyrine ,aspirin ,phenacetin, probenicid . ANTIMALARIALS chloroquine, hydroxychloroquine mepacrine, pamaquine, pentaquine, primaquine, quinine, quinocide SULFONAMIDES/SULFONES dapsone, sulfacetamide, sulfamethoxypyrimidine, sulfanilamide ,sulfapyridine sulfasalazine ,sulfisoxazole . ANTIBACTERIAL chloramphenicol, cotrimoxazole, furazolidone, nalidixic acid, nitrofurantoin nitrofurazone , para-aminosalicylic acid. MISCELLANEOUS alpha-methyldopa, ascorbic acid dimercaprol (BAL), hydralazine , methylene blue ,nalidixic acid ,naphthalene, vitamin K (water soluble) Hb S disorders (Sickle cell disorders) Defect and pathogenesis The molecular defect is the result of an abnormal autosomal gene that substitutes valine for glutamic acid in the 6th position of the β-globin chain. Under conditions of hypoxia, the Hb aggregates into insoluble long polymers that align themselves into rigid paracrystalline gels , which distort the RBC into a sickle shape. The clinical consequences of the solubility anomaly are: 1-Shortened red blood cell survival (hemolytic anemia) 2-Microvascular obstruction, which leads to tissue ischemia and infarction There are two types of Hb S disease: 1- Heterozygous state (sickle cell trait ) 2- Homozygous state (sickle cell anemia) Heterozygous state (sickle cell trait) Both Hb A and Hb S exist in individuals who have sickle cell trait; there is more Hb A than Hb S. Clinical features: Sickle cell trait is usually asymptomatic, unless the affected individual is subjected to hypoxemic stress. Otherwise, abnormalities may be limited to: 1- failure to concentrate urine. 2- painless hematuria, or both. Diagnosis: Sickle cell trait may be diagnosed by Hb electrophoresis or solubility tests (e.g slide test). It is important to detect the trait for purposes of genetic counseling. Therapy: No specific treatment is required; however, precautions to avoid hypoxemia associated with severe pneumonia, unpressurized flying, exercise at high altitudes, and G/A are in order. Tourniquet surgery and deep hypothermia should be avoided Homozygous state (sickle cell anemia) Clinical pictures: 1-In the asymptomatic period, the high levels of Hb F during fetal life and the first few months of postnatal life protect the patient. 2- Dactylitis : The earliest clinical manifestation may occur at 4 to 6 ms of age, when symmetric, painful swelling of the dorsal surfaces of the hands and feet develops. This is caused by avascular necrosis of the bone marrow of the metacarpal and metatarsal bones. During this same period, progressive anemia with jaundice and splenomegaly begins to develop. 3-Splenic sequestration crises: The spleen may suddenly become engorged with RBCs, trapping a significant portion of the blood volume. The child may presented with sudden severe pallor , severe abdominal pain and huge splenomegaly If not corrected rapidly, this can lead to hypovolemic shock and death. 4- Overwhelming infections: Pts are very susceptible to overwhelming infection, particularly with encapsulated bacteria such as pneumococci and Haemophilus influenzae; Salmonella septicemia and osteomyelitis are also seen with increased frequency in pts who have SCA. 5-Aplastic crises can occur at any age when there is suppression of erythropoiesis in response to a viral infection such as parvovirus B19 . 6-Vaso-occlusive episodes can involve any tissue. Depending on the involved organ. A vaso-occlusive episode can produce abdominal pain, bone pain, cerebrovascular accident (CVA), pulmonary infarction (acute chest syndrome) , hepatopathy, or hematuria. These episodes are often precipitated by infection, dehydration, chilling, vascular stasis, or acidosis. Repeated vaso-occlusive episodes in the spleen lead to infarction and fibrosis of this organ; it gradually regresses in size and is usually no longer palpable after the age of 5 years. 6-Late manifestations. By the time a pt reaches his late teens or early 20s, he is suffering the long-term consequences of chronic anemia, and tissue infarction. Many succumb to progressive myocardial damage with congestive heart failure. Other long-term complications include gallstones, leg ulcers, renal damage, and aseptic necrosis of the long bones. Treatment of SCA 1-Infections. Because these patients suffer from functional asplenia, the same precautions to protect them from overwhelming Gram-positive sepsis must be taken as for the patient whose spleen has been surgically removed. However, after the age of 5, there is little evidence to suggest that routine penicillin prophylaxis is required. 2-Vaso-occlusive episodes. Prevention by avoidance of dehydration, hypoxia, chilling, and acidosis. Treatment is as follows: -Analgesics should be given for pain. -When a vital organ (the brain, liver, or lung) is threatened, transfusion with packed RBCs may be necessary. -After a documented CVA, the pt remains at high risk for recurrent CVAs for an indefinite period of time; such pts should be maintained on a chronic transfusion program designed to keep the Hb S level at <30%. As is the case with chronic transfusion programs for pts who have thalassemias, iron overload may eventually necessitate chelation therapy 3-Use of agents that elevate Hb F levels. Because Hb F levels correlate inversely with disease severity, efforts have been made to identify medications that might increase Hb F levels in sickle cell anemia pts. Hydroxyurea has become the drug most commonly used for this purpose; early trials have indicated improvement in both laboratory and clinical parameters for children treated with this agent. 4- Severe aplastic crises should be treated by transfusion with packed RBCs . 5- HSC transplantation from healthy, HLA-matched sibling donors has proved to be potentially curative; however, this approach is limited by the paucity of HLA-matched siblings and the toxicity of the conditioning regimens. Antibody-mediated hemolytic anemias General considerations 1-Autoimmune hemolytic anemias (AIHA): are the result of antibodies generated by an individual's immune system against his or her own RBCs. 2- Isoimmune hemolytic anemias: result from antibodies produced by one individual against the RBCs of another individual of the same species. It can be seen in hemolytic disease of the newborn or hemolytic transfusion reactions (e.g., the transfusion of type A blood into an individual who has type B blood). Autoimmune hemolytic anemias Etiology : AIHA may be idiopathic or the result of infectious agents, drugs, lymphoid neoplasms, or disorders of immune regulation (e.g., SLE, agammaglobulinemia) Typical antibodies involved 1-Antibodies of the IgG class, for the most part, are warm reactive (i.e., they have maximal activity at 37°C). They are detected using the direct antiglobulin (Coombs) test. -These are incomplete antibodies in that they do not agglutinate RBCs, although they coat the surface. -Hemolysis occurs extravascularly . -IgG antibodies are associated clinically with autoimmune diseases, lymphomas, and viral infections. Occasionally, no underlying cause is demonstrable. 2-Antibodies of the IgM class are usually cold reactive (i.e., most have maximal activity at low temperatures). -These are complete antibodies in that they agglutinate RBCs and activate the complement sequence through C9, causing lysis of RBCs. -Hemolysis occurs intravascularly . -IgM antibodies are associated clinically with mycoplasmal pneumonia, Epstein-Barr virus, and transfusion reactions. 3-Donath-Landsteiner antibody: -It is of the IgG type, but it is exceptional in that it reacts best in the cold and can activate complement, causing hemolysis to occur intravascularly . -Its clinical associations include syphilis and viral infections. It may also be idiopathic. Therapy: depends on the cause, clinical condition of the pt, & expected duration of the illness. Because most cases of childhood AIHA are idiopathic or postinfectious and self-limited, supportive care and judicious use of transfusions and corticosteroids are the therapies most commonly used. Treatment modalities include: 1. Supportive care with bed rest and oxygen 2. Transfusion with packed RBCs 3. Corticosteroids 4. Splenectomy 5. Immunosuppressive agents