Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Pharmaceutical marketing wikipedia , lookup
Discovery and development of tubulin inhibitors wikipedia , lookup
Specialty drugs in the United States wikipedia , lookup
Polysubstance dependence wikipedia , lookup
Compounding wikipedia , lookup
Plateau principle wikipedia , lookup
Orphan drug wikipedia , lookup
Drug design wikipedia , lookup
Drug discovery wikipedia , lookup
Psychopharmacology wikipedia , lookup
Neuropharmacology wikipedia , lookup
Pharmacognosy wikipedia , lookup
Neuropsychopharmacology wikipedia , lookup
Pharmaceutical industry wikipedia , lookup
Prescription costs wikipedia , lookup
Pharmacokinetics wikipedia , lookup
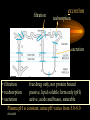
2004-2005 Module 2 #2 Pharmacokinetics 2004-2005 absorption of drugs • drugs can be given iv, im, sc, orally (po) • if given parenterally, they should be water soluble oral absorption is more complex: tablet----dissolves in gut water----lipid soluble form crosses gut barrier-----must escape liver (first pass metabolism) amount absorbed is bioavailability 2004-2005 Which definition is correct? • “Drug absorption refers to the passage of a drug from its site of administration into the circulation” –Brenner • “Absorption describes the rate and extent to which a drug leaves its site of administration and reaches its site of action” –Goodman & Gilman 2004-2005 Pharmacokinetics (how the body handles drugs) (a,d,m,e) most drugs are weak acids or bases: HA BH+ H+ + AB + H+ pH=pKa + log (base) (acid) 2004-2005 when H increases get more HA and BH+ only the non-ionized form can cross cell membranes even if the drug is not ionized, it still needs to be lipid soluble to cross cell membranes distribution of drugs once in the body, drugs distribute to tissues the ratio: amount in the body/plasma concentration is Volume of distribution (Vd) some drugs assume these: TBW = 0.6 L/Kg (42 l) Ethanol 0.54 l/kg ECF vol = 0.2 L/Kg (12 l) Gentamicin 0.31 l/kg Plasma vol. = 0.05 L/Kg (3 l) warfarin 0.14 l/kg Blood vol 2004-2005 = 5.5 l volume of distribution Cp0 Vd = Dose (mg)/Cp0 (mg/L)= Litres 2004-2005 volume of distribution-2 • sometimes the Vd is anatomically “correct” (e.g. ethanol, gentamicin, warfarin) • sometimes it’s not: (e.g.. THC 9L/Kg, amiodarone 66 L/Kg –highly lipd soluble lipid:plasma ratio >300:1) drugs with low Vd are often bound to plasma proteins drugs with high Vd are often bound to tissue components (proteins or fat) 2004-2005 Clarifying Vd • Vd is the ratio of the amount of drug in the body at any time (t) to the plasma concentration (Cp) at that time. • The only time we can be absolutely sure of the amount of drug in the body is at time 0. This is the dose of the drug that we gave iv. • We can’t measure directly the Cp0, but we can extrapolate it from later points in the log Cp vs. time curve. • When we calculate Vd, we sometimes get an unrealistic number. • We normalize Vd to Litres/Kg of body weight. If the Vd ethanol is 48 litres and the body weigh 80 Kg: the Vd 2004-2005 ethanol is 0.6 L/Kg filtration excretion reabsorption secretion • filtration free drug only, not protein bound • reabsorption passive, lipid soluble form only (pH) • secretion active, acids and bases, saturable Plasma pH is constant; urine pH varies from 5.0-8.0 2004-2005 excretion-2 if the drug is water soluble, the kidney can excrete it. the renal clearance of a drug can be calculated: Cldrug = UV = Urinary conc. UVol = ml/min P Plasma conc GFR is 125 ml/min • if the Cldrug is 125ml/min, it is filtered only • if the Cldrug is <125ml/min, it is reabsorbed • if the Cldrug is >125ml/min, it is secreted * free drug only 2004-2005 metabolism • The purpose of drug metabolism is to render drugs water soluble, so they can be excreted by the kidney • The liver is the main organ of metabolism • 2 types: ¶ phase 1: oxidation, reduction, hydrolysis ¶ phase 2: conjugation (glucuronide, sulphate) oxidation via the cytochrome P450 system is important 2004-2005 metabolism -2 reduced CYP450 picks up O2 and attaches it to the drug 2004-2005 metabolism -3 • CYP3A lots of inhibitors and inducers • CYP2D6 genetic variants (codeine)- 10% of caucasians (Chinese produce less morphine from codeine and are also less sensitive to morphine) • CYP2C9 genetic variants (warfarin – 10-20% caucasians have a variant isoform – can’t inactivate warfarin efficiently – more sensitive) many drugs use more than one CYP; saturable pathways 2004-2005 summary • pharmacokinetics describes how the body handles drugs • drugs get into and out of the body • to move from one compartment to another, the drug must be lipid soluble which means non-ionized • the kidney excretes water soluble drugs • the liver metabolizes lipid soluble drugs • CYP family is important 2004-2005 2004-2005