Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
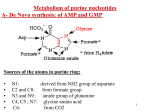
URIC ACID METABOLISM IN RELATION TO KIDNEY LEARNING OBJECTIVES By the end of this lecture the student should know: • 1. Uric acid • 2. Uric acid pathway • 3. Hypouricemia • 4. Hyperuricemia • 5. Gout URIC ACID • • • • • The final breakdown product of purine degradation. Urates, the ionized forms of uric acid, predominate in plasma extracellular fluid and synovial fluid, as monosodium urate at pH 7.4. Plasma is saturated with monosodium urate. At higher concentrations, plasma supersaturated, creating the potential for urate crystal precipitation. The pH of urine greatly influences the solubility of uric acid. URIC ACID • • • Ionized forms of uric acid in urine include mono- and disodium, potassium, ammonium, and calcium urates. Purine nucleotides are synthesized and degraded in all tissues, urate is produced only in tissues that contain xanthine oxidase, primarily the liver and small intestine. Urate production varies with the purine content of the diet and the rates of purine biosynthesis, degradation, and salvage. URIC ACID • • • Daily synthesis 400mg Dietary sources 300mg Normal uric acid pool: 1200mg in males and 600mg in females 75% excreted in urine, remainder in GIT where it’s degraded to allantoin by bacterial enzymes. • • Men: 3.4–8 mg/dL Women: 2.4–6 mg/dL Collection: Tiger top or red top tube Increase associated with increased catabolism, nucleoprotein synthesis, or decreased renal clearing of uric acid (ie, thiazide diuretics, renal failure). PURINE BIOSYNTHESIS PURINE CATABOLISM PATHWAY URIC ACID The kidneys clear urate from the plasma and maintain physiologic balance by utilizing specific organic anion transporters (OATs) including urate transporter 1 (URAT1) and human uric acid transporter (hUAT). Four-component model has been used to describe the renal handling of urate/uric acid: (1) Glomerular filtration, (2) Tubular reabsorption, (3) Secretion, (4) Postsecretory reabsorption. • It is now apparent that they are carried out in parallel by these transporters. • Uricosuric compounds directly inhibit URAT1, In contrast, antiuricosuric compounds (those that promote hyperuricemia), such as nicotinate, pyrazinoate, lactate, and other aromatic organic acids, serve as the exchange anion inside the cell, thereby stimulating anion exchange and urate reabsorption HYPOURICEMIA Defined as a serum urate concentration <120 mol/L (2.0 mg/dL) can result from decreased production of urate, increased excretion of uric acid, or a combination of both mechanisms. Causes: • • • Hepatocellular disease with reduced purine synthesis Defective tubular reabsorption Over treatment • Deficiency of xanthine oxidase( xanthinuria also present) Most hypouricemia results from increased renal uric acid excretion. • Other causes include neoplastic disease, hepatic cirrhosis, diabetes mellitus, and inappropriate secretion of vasopressin; Defects in renal tubular transport such as primary Fanconi syndrome and heavy metal toxicity; Hypouricemia can be familial. Some of these cases result from a lossof-function mutation in SLC22A12, the gene that encodes for URAT1. • • HYPOURICEMIA • • • • Idiopathic Familial juvenile gouty nephropathy: This is a rare autosomal dominant condition characterized by progressive renal insufficiency. These patients have a low fractional excretion of urate (typically 4%). Kidney biopsy findings indicate glomerulosclerosis and tubulointerstitial disease but no uric acid deposition. HYPERURICEMIA • • • • Defined as a plasma urate concentration >408 mol/L (6.8 mg/dL). Can result from increased production or decreased excretion of uric acid or from a combination of the two processes. Sustained hyperuricemia predisposes some individuals to develop clinical manifestations including gouty arthritis , urolithiasis, and renal dysfunction Hyperuricemia is present in between 2.0 and 13.2% of ambulatory adults and is even more frequent in hospitalized individuals. CAUSES OF HYPERURICEMIA • May be classified as primary or secondary depending on whether the cause is innate or is the result of an acquired disorder. CAUSES OF HYPERURICEMIA 1. Elevated 5-phosphoribosyl-1-pyrophosphate synthetase (PRPP synthetase) activity. PRPP synthetase is responsible for the synthesis of PRPP (activated ribose) necessary for de novo synthesis of purine nucleotides 2. Hypoxanthine-guanine phosphoribosyl transferase (HGPRT) deficiency. HGPRT enzyme involved in salvage of purine nucleotides 3.Decrease salvage leads to increased metabolism 4. increased production of uric acid HGPRT, PRPP synthetase . RENAL DYSFUNCTION THAT RESULTS IN DECREASED IN URIC ACID EXCRETION • • • • Uric Acid Transporter (URAT1) : Organic anion transporter (OAT). Uric acid reabsorption URAT 1 exchange uric acid with endogenous and exogenous anions : e.g Lactic acid, Butyric acid, Nicotinic acid, PZA- Polymorphisms or mutations of the URAT1 • Uricosuric drugs: Probenecid, Benzbromarone, Sulfinpyrazone, and losartan inhibit the uptake of uric acid by inhibiting URAT 1 RENAL INSUFFICIENCY • • Renal failure is one of the more common causes of hyperuricemia. In certain renal disorders, such as medullary cystic disease and chronic lead nephropathy, hyperuricemia is commonly observed even with minimal renal insufficiency. Syndrome X: This metabolic syndrome is characterized by hypertension, obesity, insulin resistance, dyslipidemia, and hyperuricemia. This is associated with a decreased fractional excretion of urate by the kidneys. DRUGS • • • • • Causative drugs include diuretics, low-dose salicylate, cyclosporine, pyrazinamide, ethambutol, nicotinic acid, and methoxyflurane. Hypertension Acidosis: Types that cause hyperuricemia include lactic acidosis, diabetic ketoacidosis, alcoholic ketoacidosis, and starvation ketoacidosis. Preeclampsia and eclampsia: The elevated uric acid associated with these conditions is a key clue to the diagnosis because uric acid levels are lower than normal in healthy pregnancies. Hypothyroidism Hyperparathyroidism Sarcoidosis Lead intoxication (chronic): Trisomy 21 OVERPRODUCTION • Idiopathic HGPRT(hypoxanthine-guanine phosphoribosyl transferase) deficiency (Lesch-Nyhan syndrome):This is an inherited X-linked disorder. • HGRPT catalyzes the conversion of hypoxanthine to inosinic acid, in which PRPP serves as the phosphate donor. The deficiency of HGPRT results in accumulation of PRPP, which accelerates purine biosynthesis with a resultant increase in uric acid production. • In addition to gout and uric acid nephrolithiasis, these patients develop a neurologic disorder that is characterized by choreoathetosis, spasticity, growth, mental function retardation, and, occasionally, self-mutilation. • • • • • • • • • • • Partial deficiency of HGPRT (Kelley-Seegmiller syndrome): This is also an X-linked disorder. Patients typically develop gouty arthritis in the second or third decade of life, have a high incidence of uric acid nephrolithiasis, and may have mild neurologic deficits. Increased activity of PRPP synthetase: This is a rare X-linked disorder in which patients make mutated PRPP synthetase enzymes with increased activity. These patients develop gout when aged 15-30 years and have a high incidence of uric acid renal stones. Purine-rich diet: A diet rich in meats, organ foods, alcohol, and legumes can result in an overproduction of uric acid. Increased nucleic acid turnover: This may be observed in persons with hemolytic anemia and hematologic malignancies such as lymphoma, myeloma, or leukemia. Tumor lysis syndrome: This may produce the most serious complications of hyperuricemia. Glycogenoses III, V, and VII Ethanol increases the production of uric acid by causing increased turnover of adenine nucleotides. It also decreases uric acid excretion by the kidneys, which is partially due to the production of lactic acid. Exercise: Exercise may result in enhanced tissue breakdown and decreased renal excretion due to mild volume depletion. Deficiency of aldolase B (fructose-1-phosphate aldolase): This is a fairly common inherited disorder, often resulting in gout. Glucose-6-phosphatase deficiency (glycogenosis type I, von Gierke disease): This is an autosomal recessive disorder characterized by the development of symptomatic hypoglycemia and hepatomegaly within the first 12 months of life. • Additional findings include short stature, delayed adolescence, enlarged kidneys, hepatic adenoma, hyperuricemia, hyperlipidemia, and increased serum lactate levels FRUCTOSE INDUCES AN INCREASE IN URIC ACID : Fructose rapidly raises uric acid as a consequence of activation of fructokinase with ATP consumption, resulting in intracellular phosphate depletion. • AMP deaminase activity is stimulated by low levels of phosphorus, resulting in greater degradation of AMP to uric acid • Fructose also competes for uric acid excretion. GOUT ARTHRITIS: • • • • Classic example of crystal-induced inflammation of synovial joints. It is a common condition, presenting in 1–4% of adult men. precipitation of monosodium urate in tissues and joints eliciting an intense inflammatory response. Treatment with allopurinol( xanthine oxidase inhibitor and decrease PRPP concentration). Urate stone ( acidic urine, hyperuricemia.) GOUT • • • • Elevated uric acid levels in the blood Uric acid crystals will form in the extremities with a surrounding area of inflammation. This is called a tophus and is often described as an arthritic “great toe”. Can be caused by a defect in an enzyme of purine metabolism or by reduced secretion of uric acid into the urinary tract. Deposition of monosodium urate crystals in the joint space leads to episodes of severe acute joint pain and swelling (particularly in the great toe, midfoot, ankle, and knee). These episodes tend to resolve completely and spontaneously within a week even without treatment. GOUTY ARTHRITIS • • • Crystals of monosodium urate free in the joint activate a number of inflammatory pathways. The crystals are coated with immunoglobin G (IgG), which activates complement. Similarly, the kallikrein system is activated. As a result, there is vasodilatation and influx of neutrophils. Phagocytosed uric acid crystals stimulate neutrophils to release prostaglandins and lysosomal enzymes and induce oxidant production. GOUTY ARTHRITIS • • Further, neutrophils are unable to digest the phagocytosed urate crystal, which ultimately results in lysosomal rupture, cell death, and spillage of cellular contents extracellularly. Simultaneously, free uric acid crystals activate mononuclear cells and synoviocytes with the production of inflammatory cytokines, including interleukin (IL)-1 and tumor necrosis factor (TNF)-alpha, which account for some of the systemic symptoms seen particularly in recurrent attacks of gout. Factors posited as reasons for involvement of the lower extremity • Uric acid solubility is temperature dependent and thus colder peripheral joints would be less likely to maintain its solubility. Further, hyaluronic acid, a major component of articular cartilage, increases the solubility of uric acid. Finally, it has been suggested that, with ambulation, the simple trauma causes fluid to enter the joint. At night, water is more rapidly absorbed from the joint than uric acid, increasing the concentration of uric acid, thus potentially causing its precipitation and triggering an attack. • REFERENCES • • • Lippincott’s Biochemistry Biochemistry by Chatterji Harpers Biochemistry THANK YOU