Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Nitric Oxide and Sepsis, Hemodynamic Collapse, and the Search for Therapeutic Options
Larry H Bernstein, MD, FCAP, Curator, Reporter, EAW, Pharmaceutical Intelligence
This document explores the current understanding of sepsis as a cascade of events that involves the
microcirculation unevenly because of a differential effect on the large and contiguous intestinal
epithelium, secondary effects on cardiopulmonary blood flows and cardiac output, and the role of Nitric
Oxide in the emergence of beneficial and potentially deleterious effects. This leads to a substantial body
of work on therapeutic targets, either aimed at total inhibition or selective inhibition of NO synthase,
and the special role of iNOS. This is another of a series of discussions on the metabolic and regulatory
role of NO in health and disease.
Introduction
Antioxidants are essential, and are involved in several important biological processes such as immunity,
protection against tissue damage, reproduction, growth and development. Antioxidants preserve
adequate function of cells against homeostatic disturbances such as those caused by septic shock, aging
and, in general, processes involving oxidative stress. This review focuses on the involvement of reactive
oxygen and nitrogen species.
The presence of free radicals in biological materials was discovered about 50 years ago. Today, there is a
large body of evidence indicating that patients in hospital intensive care units (ICUs) are exposed to
excessive free radicals from drugs and other substances that alter cellular reduction -oxidation (redox)
balance, and disrupt normal biological functions. However, low levels of free radicals are also vital for
many cell signaling events and are essential for proper cell function.
Excess free radicals can result from a variety of conditions such as tissue damage and hypoxia (limiting
oxygen levels), overexposure to environmental factors (tobacco smoke, ultraviolet radiation, and
pollutants), a lack of antioxidants, or destruction of free radical scavengers. When the production of
damaging free radicals exceeds the capacity of the body's antioxidant defenses to detoxify them, a
condition known as oxidative stress occurs.
Free Radicals and Antioxidants: an Overview
A free radical can be described as any atom or a group of atoms or molecules in which there is at least
one unpaired electron in the outermost shell . These free radicals are very reactive with adjacent
molecules such as lipids, proteins, and carbohydrates and can cause cellular damage. Paradoxically, free
radicals can also be produced by many cells as a protective mechanism, for example neutrophils
produce free radicals to attack and destroy pathogens, while the liver uses free radicals for
detoxification. However, the presence of free radicals within the body can also have a significant role in
the development and progression of many disease processes for example heart disease, hypertension,
cerebrovascular accidents, and diabetic complications. Any free radical involving O2 is referred to as a
reactive oxygen species (ROS).
Normal cellular metabolism involves the production of ROS, and in humans, superoxide (O2 -) is the
most commonly produced free radical. Phagocytic cells such as macrophages and neutrophils are
prominent sources of O2 -. During an inflammatory response, these cells generate free radicals that
attack invading pathogens such as bacteria and, because of this, the production of O2- by activated
phagocytic cells in response to inflammation is one of the most studied free radical producing systems.
The majority of the H2O2 is broken down to O2 and water by the antioxidant enzyme catalase. In
addition to catalase, glutathione peroxidase can also break down H2O2 and also any peroxides that form
on lipids within the body [5]. When O2 - reacts with nitric oxide (NO), the toxic product peroxynitrite
(ONOO-) is formed.
Cellular ROS originate from O2- generated as a by-product of oxidative phosphorylation (mitochondrial
respiration), they differ in their mechanism of production, necessary cofactors, diffusion range,
hydrophobicity, biological targets, detoxification pathways and breakdown products. O-2 damaging
reactions largely involve disassembly of iron-sulphur clusters in proteins. H2O2 or O-2 alone lacked
reactivity toward iron regulatory protein-1 (IRP-1), but a combined action of the two species induced
reversible inactivation of IRP-1. Such an effect was attributed to direct interactions of O-2 and H2O2
with a preformed pool of IRP-1, resulting in reversible modifications of -SH residues; in fact, its action
would be limited to removing only iron atoms, an effect sufficient to abolish enzyme activity.
The hydroxyl radical (.OH) is the most reactive of the free radical molecules. OH- damages cell
membranes and lipoproteins by a process termed lipid peroxidation. In fact, lipid peroxidation can be
defined as the process whereby free radicals “steal” electrons from the lipids in our cell membranes,
resulting in cell damage and increased production of ROS. This process takes place in 3 stages:
1. Initiation: In a peroxide-free lipid system, the initiation of a peroxidation sequence refers to the
attack of an ROS (with sufficient reactivity) able to abstract a hydrogen (H) atom from a
methylene group (- CH2-).
2. 2. Propagation: A peroxyl radical is able to abstract H from another lipid molecule (adjacent fatty
acid), especially in the presence of metals such as copper or iron, thus causing an autocatalytic
chain reaction. The peroxyl radical combines with H to give a lipid hydroperoxide (or peroxide).
3. Termination: formation of a hydroperoxide. Lipid peroxidative damage to lipids in low-density
lipoprotein (LDL) plays an important role in atherosclerosis [9]. To protect against oxidative
damage, organisms have developed a variety of antioxidant defenses that include proteins,
compounds such as vitamins, and specialized antioxidant enzymes.
Lipid-soluble antioxidants are located in the cellular membranes and lipoproteins, whereas the watersoluble antioxidants are present in the aqueous environments, such as fluids inside cells and in the
blood. Preventative antioxidant enzymes inside the cell are an important defense against free radicals.
In humans, the highest levels of SOD are found in the liver, adrenal gland, kidney, and spleen [10].
Catalase and glutathione peroxidase both work to detoxify O2-reactive radicals by catalyzing the
formation of H2O2 derived from O2 -. The liver, kidney, and red blood cells possess high levels of
catalase, which helps to detoxify chemicals in the body. The water-soluble tripeptide-thiol glutathione
also plays an important role in a variety of detoxification processes. Glutathione is found in millimolar
concentrations in the cell cytosol and other aqueous phases, and readily interacts with free radicals,
especially the hydroxyl radical, by donating a hydrogen atom.
Sepsis and Signaling Pathways
Serious infections trigger systemic inflammatory response and can result in sepsis. It is believed that
sepsis and therefore septic shock are due to the inappropriate increase in the innate immune response
via circulating and tissue inflammatory cells, such as monocytes/macrophages and neutrophils. These
cells normally exist in a nonactivated state but are rapidly activated in response to bacteria. Sepsis
induces a dysfunction in immune cells that contributes to the development of injuries by producing
mediators such as cytokines and ROS.
Lipopolysaccharide (Lps) Signaling
LPS of Gram-negative organisms induces macrophages to secrete cytokines, which in turn activate T, and
B cells to upregulate the adaptive immune responses. Toll-like receptor 4 (TLR4) is the LPS receptor and
its stimulation induces nuclear factor kB (NF-kB) activation. The activation of NF-kB involves
phosphorylation and degradation of IkB, an inhibitor of NF-kB. The NF-kB/IkB system exerts
transcriptional regulation on proinflammatory genes encoded for various adhesion molecules and
cytokines. Activation of NF-kB leads to the induction of NF-kB binding elements in their promoter
regions and also leads to the induction of NF-kB dependent effector genes, which produce modifications
in blood flow, and aggregation of neutrophils, and platelets. This results in damaged endothelium and
also coagulation abnormalities often seen in patients with sepsis and septic shock. Therefore, NF-kB is
reported to be an O2 sensor in LPS-induced endotoxemia.
Free Radicals and Antioxidants In Sepsis
The sources of ROS during sepsis are:
(1)
(2)
(3)
(4)
the mitochondrial respiratory chain.
the metabolic cascade of arachidonic acid.
the protease-mediated enzyme xanthine oxidase.
granulocytes and other phagocytes activated by complement, bacteria, endotoxin, lysosomal
enzymes, etc.
(5) Other oxidases mainly NADPH oxidase.
Under normal physiological conditions, the majority of ROS are formed during cellular respiration and by
activated phagocytic cells, including neutrophils, involved in the inflammatory response. ROS have
physiologically essential roles in mitochondrial respiration, prostaglandin production pathways and host
defense . The electron reduction of O2 occurs in the mitochondrial electron transport system of all
aerobically respiring cells. The enzyme catalyzing this transition metals iron and copper in its active site.
These ions can be paramagnetic and contain stable unpaired electrons. By using the unpaired electrons
in these transition metals to control the O2 reactions, mitochondria prevent the unwanted release of
ROS.
In sepsis, there are several potential sources of ROS, including the mitochondrial respiratory electron
transport chain, xanthine oxidase activation as a result of ischemia and reperfusion, the respiratory
burst associated with immune cell activation, arachidonic acid metabolism and NADPH oxidase. In fact,
activated immune cells produce O2 - as a cytotoxic agent as part of the respiratory burst via the action
of membrane-bound NADPH oxidase on O2. The increase of ROS after LPS challenge has been
demonstrated in different models of septic shock in peritoneal macrophages and lymphocytes.
This disturbance in the balance between pro-oxidants (ROS) and antioxidants in favor of the former is
characteristic of oxidative stress in immune cells in response to endotoxin. In this context, a typical
behavior of these cells under an oxidative stress situation implies changes in different immune
functions such as an increase in adherence and phagocytosis and a decrease in chemotaxis . Neutrophils
play a crucial role in the primary immune defense against infectious agents,which includes phagocytosis
and the production of ROS.
Antioxidant Defenses
Antioxidants are central to the redox balance in the human body. They do not act in isolation, but
synergistically with other classes of molecules. Primary antioxidants prevent oxygen radical formation,
by either removing free radical precursors or by inhibiting catalysis, e.g. the enzymes glutathione
peroxidase and catalase. Secondary antioxidants react with ROS which have already been formed, either
to remove or inhibit them, e.g. vitamins C and E. Endogenous antioxidant defenses exist in a number of
locations, namely intracellularly, on the cell membrane and extracellularly. The immune system is highly
reliant on accurate cell-cell communication for optimal function, and any damage to the signaling
systems involved will result in an impaired immune responsiveness. Oxidant-mediated tissue injury is a
particular hazard to the immune system, since phagocyte cells produce ROS as part of the defense
against infection. Therefore, adequate amounts of neutralizing antioxidants are required to prevent
damage to the immune cells themselves.
The SOD enzymes are a family of metalloenzymes which rapidly promote the conversion of O2- to H2O2.
Three forms of SOD are recognized to be important: copper-zinc SOD (cytoplasmic-located), manganese
SOD (mitochondrial-located) and extracellular SOD (extracellular matrix-located). Catalase and
glutathione peroxidase, a selenium containig enzyme which requires the presence of reduced GSH for its
action, both catalyze the conversion of H2O2 to H 2O. GSH also has direct antioxidant activity, through
donation of hydrogen ions, to repair damaged DNA. Oxidative stress and modulation on GSH/GSSG
(GSSG=oxidized GSH) levels also up-regulate gene expression of several other antioxidant proteins, such
as manganese SOD, glutathione peroxidase, thioredoxin (Trx) and metallothionein.
Effects of Nitric Oxide
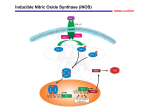
NO is synthesized from L-arginine by different isoenzymes of (NOS), and is implicated in a wide range of
disease processes, exerting both detrimental and beneficial effects at the cellular and vascular levels. To
date, three main isoforms of NOS are known:
(1) neuronal NOS (NOS-1 or nNOS),
(2) inducible NOS (NOS-2 or iNOS), and
(3) endothelial NOS (NOS-3 or eNOS).
NO has been shown to play a key role in the pathogenesis of septic shock. Hyperproduction of NO
induces excessive vasodilation, changes in vascular permeability, and inhibition of noradrenergic nerve
transmission, all characteristics of human septic shock.
The recogniton of NO production by activated macrophages as part of the inflammatory process was an
important milestone for assesing both the biological production of NO and the phenomenon of
induction of NOS activity. The observation has been extended to neutrophils, lymphocytes, and other
cell types. The role of NO in the pathophysiology of endotoxic shock was advanced by Thiemermann and
Vane, who observed that administration of the specific NOS inhibitor N-methyl-L-arginine (L-NMMA)
decreased the severe hypotension produced by administration of LPS. Other groups simultaneously
reported similar results indicating that endotoxin increases NO production and prompted the idea that
pharmacological inhibition of NOS may be useful in the treatment of inflammation and septic shock.
However, clinical trials using L-NMMA failed to show a beneficial effect in septic shock patient. The
major limitation for the use of NOS inhibitors in clinical studies is the development of pulmonary
hypertension as a side effect of NOS blockade, which can be alleviated by the use of inhaled NO.
However, several compounds which modulate NO synthesis have been patented in recent years, such as
various inflammatory mediators that have been implicated in the induction and activation of iNOS,
particularly IFNg, TNFa, IL-1b, and platelet-activating factor (PAF) alone or synergistically. In addition to
the activation of iNOS, cytokines and endotoxin may increase NO release by increasing arginine
availability through the opening of the specific y+ channels and the expression of the cationic amino
acid transporter (CAT), or by increasing tetrahydrobiopterin levels, a key cofactor in NO synthesis.
Several experimental studies have demonstrated a decrease in NOS activity resulting in an impairment
in endothelial-dependent relaxation during endotoxemia and experimental sepsis, possibly as the result
of a cytokine-or hypoxia-induced shortened half-life of NOS mRNA, or of altered calcium mobilization.
NO exerts in vitro toxic effects including nuclear damage, protein and membrane phospholipid
alterations, and the inhibition of mitochondrial respiration in several cell types. Mitochondrial
impairment could also be considered as an adaptive phenomenon, decreasing cellular metabolism when
the energy supply is limited. The toxicity of NO itself may be enhanced by the formation of ONOO- from
the reaction of NO with O-2. Therefore, the multiple organ failure syndrome (MOFS) that often
accompanies severe sepsis may be related to the cellular effects of excess NO or ONOO-.
Involvement of Nitrogen Species
NO reacts rapidly with ferrous iron, and at physiological concentrations, NO also binds to soluble
guanylate cyclase and to another hemoprotein, cytochrome c oxidase (Complex IV), the terminal
enzyme of the mitochondrial respiratory chain. NO can therefore control cellular functions via the
reversible inhibition of respiration. There are a number of reactive NO species, such as N2O3 and
ONOO- that can also alter critical cellular components.
During the first hours after injury, iNOS-mediated NO production is upregulated, producing a burst of
NO that far exceeds basal levels. This overabundance of NO produces significant cellular injury via
several mechanisms.
(1) NO may directly promote overwhelming peripheral vasodilation, resulting in vascular
decomposition;
(2) NO may upregulate the transcription NF-kB initiating an inflammatory signaling pathway that, in
turn, triggers numerous inflammatory cytokines.
(3) NO also interacts with the O-2 to yield ONOO-, a highly reactive compound that exacerbates the
injury produced by either O-2 alone or NO alone.
The ONOO- generation which occurs during fluid resuscitation in the injured subject produces cellular
death by enhancing DNA single strand breakage, activates the nuclear enzyme polyADP ribose
synthetase (PARS), leading to cellular energy depletion and cellular necrosis. The detrimental effects of
ONOO- in shock and resuscitation have been attributed to oxidation of sulfhydryl groups, the nitration
of tyrosine, tryptophane, and guanine, as well as inhibition of the membrane sodium-potassium
adenosine triphosphatase. PARS activation depletes NAD and thus alters electron transport, ATP
synthesis, and glycolysis; and leads to DNA fragmentation and cellular apoptosis.
The activation of monocytes, macrophages and endothelial cells by LPS results in the expression of iNOS,
and consequently increases the transformation of L-arginine to NO, which can combine with O2- to form
ONOO-, causing tissue injury during shock, inflammation and ischemia reperfusion. NO stimulates H2O2
and O-2 production by mitochondria, increasing leakage of electrons from the respiratory chain. H2O2,
in turn, participates in the upregulation of iNOS expression via NFkB activation. ONOO- has been shown
to stimulate H2O2 production by isolated mitochondria. On the other hand, NO can decrease ROSproduced damage that occurs at physiological levels of NO. The high reactivity of NO with radicals might
be beneficial in vivo by scavenging peroxyl radicals and inhibiting peroxidation. ONOO- may also be a
signal transmitter and can mediate vasorelaxation, similarly to NO.
Local generation of RNS contributes to tissue injury. Recent studies have demonstrated that activation
of the nuclear enzyme poly(ADP-ribose) polymerase-1 by RNS-mediated DNA damage is an important
pathway of tissue injury in conditions associated with oxidative stress. Increased formation of RNS in
response to endotoxin challenge is organ specific.
In sepsis, NO may exert direct and indirect effects on cardiac function. Sustained generation of NO
occurs in systemic inflammatory reactions, such as septic shock with involvement in circulatory failure.
In fact, myocardial iNOS activity has been reported in response to endotoxin and cytokines and inversely
correlated with myocardial performance. Low-to-moderate doses of iNOS inhibitors restore myocardial
contractility in hearts exposed to proinflammatory cytokines, whereas at higher doses, the effects are
reversed. This finding may indicate that small amounts of NO produced by iNOS may be necessary to
maintain contractility and can be cardio-protective in experimental sepsis.
Nitric oxide in Septic Shock
A list of effects of NO in sepsis is as follows.
(1)
(2)
(3)
(4)
(5)
(6)
(7)
(8)
Inhibition of nitric oxide synthesis causes myocardial ischemia in endotoxemic rats
Nitric oxide causes dysfunction of coronary autoregulation in endotoxemic rats
Prolonged inhibition of nitric oxide synthesis in severe septic shock
Effect of L-NAME, an inhibitor of nitric oxide synthesis, on cardiopulmonary function in human
septic shock
Pulmonary hypertension and reduced cardiac output during inhibition of nitric oxide synthesis
in human septic shock
Effect of L-NAME, an inhibitor of nitric oxide synthesis, on plasma levels of IL-6, IL-8, TNF-u and
nitrite/nitrate in human septic shock
Endothelin-1 and blood pressure after inhibition of nitric oxide synthesis in human septic shock
Distribution and metabolism of NO-nitro-L-arginine methyl ester in patients with septic shock
The possible involvement of the L-arginine-NO pathway in both the vascular and cellular processes seen
in sepsis has been supported by numerous in vitro and in vivo studies. iNOS appears to be expressed in a
wide array of cell types during sepsis, including immune cells (such as macrophages, neutrophils, T
lymphocytes), as well as cells outside the classical immune system (for example, hepatocytes, Kuppfer
cells, vascular smooth muscle cells, endothelial cells, and fibroblasts). Expression of iNOS is regulated,
both positively and negatively, by a number of mediators present during infection and inflammation.
The main stimuli for iNOS induction indude lipopolysaccharide (LPS), interferon-y, interleukin (IL)-10,
and tumor necrosis factor (TNF)-a; inhibitory cytokines, such as transforming growth factor-5, IL-4 and
IL-10, as well as glucocorticoids, can prevent this induction. The expression of iNOS in response to these
agents differs among cell types, but a maximal inducing effect is generally obtained by the combination
of microbial products and cytokines acting synergistically. iNOS activity is also regulated by substrate
and cofactor availability. Tetrahydrobiopterin (BH4), an essential cofactor for the enzyme, is coinduced
with iNOS in cytokine-stimulated vascular smooth muscle cells.
NO is a simple molecule, but its widespread production in sepsis, coupled with its effects on a variety of
intracellular and extracellular target molecules, results in a complex array of biologic roles. Interaction of
NO with the metalloproteins in a number of key enzymes can modulate their activity. Many of the
signaling actions of 'NO are mediated by soluble guanylate cyclase. By binding the iron on the heme
component of soluble guanylate cyclase, NO is able to activate the enzyme leading to cyclic guanosine
monophosphate (cGMP) formation. Increased cGMP levels account for several of the important cellular
actions of NO, including smooth muscle relaxation, platelet aggregation and adherence, as well as
neutrophil chemotaxis. However, *NO can adversely affect cellular metabolism through its disruption of
iron-sulfur clusters in essential energy-generating enzymes involved in mitochondrial electron transport,
glycolysis, and the Krebs cycle. Further, high concentrations of induced macrophage produced NO can
directly interfere with DNA in target cells, resulting in fragmentation.
Another critical reaction that 'NO undergoes during inflammation is with the superoxide anion radical
(02j, yielding peroxynitrite (OONO-). OONO- is a potent oxidant that can decay under acidic conditions
to produce a powerful hydoxyl-like free radical. This reaction between *NO and 02 can have a protective
or damaging consequence, depending on the individual sites and rates of production of the free radicals,
and the redox status of both the generating cells as well as the target cells. OONO- formation can
initiate adverse effects such as lipid peroxidation of membranes, and modification of structural proteins
through nitration of tyrosine residues (14). Indeed, increased levels of 3-nitrotyrosine have been
detected in the lungs of patients with sepsis and animals with acute lung injury. However, OONO- can
also S-nitrosylate glutathione and other thiol-containing substances to form S-nitrosothiols, which have
marked cardioprotective and cytoprotective effects.
The damaging effect of NOS inhibition may be, in part, mediated by oxygen radicals and platelet
deposition, suggesting a cytoprotective role of NO in preventing microvascular thrombosis and as a free
radical scavenger. In addition, 'NO has a protective role in hepatic microcirculatory dysfunction during
sepsis through its effect on leukocyte adherence to sinusoidal walls. 'NO may also protect against
circulatory vasoconstrictors during inflammation, as enhanced 'NO synthesis counteracted
phenylephrine-induced increases in intrahepatic resistance in endotoxin-treated rats. Finally, we have
recently demonstrated that different types of NOS inhibitors resulted in detectable apoptosis in the liver
following LPS injection. This increase in apoptosis was present even with L-N-iminoethyl-lysine (L-NIL), a
rather specific inhibitor of iNOS, revealing another important protective role of NO as an antiapoptotic
agent in sepsis.
Even though overproduction of *NO in the vasculature contributes to the vasodilatation seen in septic
shock, iNOS expression during inflammation also represents a beneficial, adaptive response in some
organ systems. Moreover, different tissues can react dissimilarly to the effects of 'NO cytotoxicity. In this
setting, global nonselective inhibition of NOS, including the potentially undesirable consequences of
eNOS inhibition, would be harmful. If confirmed, this would suggest that use of isoform-specific
inhibitors of NOS within the vascular bed would be more appropriate.
Pulmonary Hypertension and Reduced Cardiac Output
Pulmonary hypertension and reduced cardiac output can be major side effects of continuous NO
synthase inhibition. Pulmonary vasoconstriction is undesirable because it may compromise pulmonary
gas exchange and because it increases the workload on the right ventricle. In cases where strain already
exists on the right ventricle (e.g. sepsis or PEEP ventilation) or in cases where right sided cardiac reserve
is minimal, such increase in workload may lead to right ventricular failure, reduced cardiac output and
compromised tissue perfusion.
Blood pressure and systemic vascular resistance increased during infusion of the NO synthase inhibitor
L-NAME, and the dosage of catecholamines was reduced. The vasoconstrictive response to L-NAME
most likely was the result of blocking the NO system . In addition to the systemic effects of L-NAME,
severe pulmonary vasoconstriction was observed with L-NAME. Analogous to these findings, in patients
with Adult Respiratory Distress Syndrome (ARDS), inhalation of NO is reported to be beneficial by
causing local vasodilation in bronchial and pulmonary circulation which results in reduced pulmonary
vascular resistance and improved oxygenation. This suggests that the pulmonary circulation is sensitive
to the vasodilating effects of both endogenous and exogenous NO. Pulmonary vasoconstriction is not,
therefore, unexpected with systemic inhibition of NO synthesis. With a continuous infusion of L-NAME,
pulmonary vascular resistance increased five-fold, whereas systemic vascular resistance "only" doubled.
pulmonary hypertension was reversible after stopping L-NAME infusion. In prior experiments with a
lower dose of LNAME, pulmonary vasoconstriction was less pronounced and did not result in pulmonary
hypertension.' Thus, pulmonary hypertension is a dose-related effect of L-NAME that can probably be
attributed to overdosing of the drug. Reduced cardiac output may have directly resulted from the
extreme increase in pulmonary vascular resistance compromising venous return and left ventricular
preload and/or a reflex reduction in heart rate by the increase in vascular resistance and blood pressure.
S-methyl-isothiourea, a relatively selective inhibitor of iNOS activity, decreased pulmonary leak and
improved survival in endotoxemia. However, because of the tissue-protective and antiapoptotic effects
of NO, even selective iNOS inhibitors may be detrimental in certain tissues during sepsis.Combining the
salutary effects of site-specific local donors that exploit the cytoprotective actions of 'NO with specific
agents that combat the deleterious hypotensive and tissue-damaging effects of 'NO overproduction may
be needed to treat septic shock. In this regard, inhaled 'NO gas has shown promise as a selective
pulmonary vasodilator.
Role of Nitric Oxide in Inflammation and Tissue Injury
Since the discovery that nitric oxide ('NO) accounts for the biologic activity of endothelial-derived
relaxing factor, a torrent of research over the last decade has focused on its role, protective or
detrimental, in myriad pathophysiologic conditions. Recently, increasing attention has focused on 'NO as
a possible mediator of the severe hypotension and impaired vasoreactivity characteristic of circulatory
failure. Experimental and clinical studies have suggested NOS inhibition might have therapeutic
potential in circulatory shock, and other studies have demonstrated the beneficial nature of iNOS
expression in modulating tissue perfusion and mediating cytotoxicity. However, inhibition of 'NO
synthesis in experimental and clinical studies of shock has yielded mixed, sometimes contradictory,
results. Overproduction of 'NO in the vasculature may result in systemic vasodilatation, but still 'NO
synthesis has a beneficial role in regulating organ perfusion and mediating cytotoxicity.
diminished *NO production occurs with hemorrhage
These findings are consistent with those in trauma patients, where nitrite and nitrate levels were
reduced for prolonged periods after injury. This impairment of 'NO production in victims of hemorrhagic
hypotension may be due to impairment of eNOS, and indeed, several investigators have demonstrated
decreased vasodilatory activity in vascular rings taken from hemorrhaged animals in response to
agonists that stimulate endothelial 'NO production. In studies of hemorrhagic shock no iNOS expression
could be detected until the very late irreversible phase of HS. The hemodynamic instability associated
with decompensation occurred well before NOS induction.
Using either the selective inhibitor L-NIL or iNOS knockout mice, we found that iNOS inhibition or
deficiency not only prevented the upregulation of the inflammatory cytokines IL-6 and granulocyte
colony-stimulating factor following resuscitation from HS but also produced a marked reduction in lung
and liver injury. Furthermore, the activation of the proinflammatory transcriptional factors nuclear
factor kappa B and signal transducer and activator of transcription 3 was also reduced, suggesting iNOS
upregulation has a key role in proinflammatory signaling and the subsequent activation of inflammatory
cascades. Recent studies have implicated a possible redox-sensitive mechanism. 'NO activates the
critical signaling enzyme p21ras through S-nitrosylation.
Vascular quenching of 'NO using scavengers may again provide an alternative to NOS inhibition as a
means to achieve the goal of reducing 'NO levels. Use of 'NO scavengers after HS and resuscitation may
serve to supplement a possibly depleted antioxidant defense system and limit the harmful effects of
free radicals such as OONO- and hydoxyl radicals. Removal of 'NO by this method is complicated by the
extreme rapidity of the reaction between 'NO and 02'- .
iNOS upregulation also has a beneficial protective role in several organ systems. In conditions where
excess NO production results in maladaptive damaging consequences with disruption of homeostasis,
the therapeutic strategy should be to remove this surplus 'NO without adversely affecting the
cytoprotective actions of *NO. Interfering with the physiologic and microcirculatory role of eNOS
through nonselective, global inhibition of NOS is undesirable in shock.
Effects of nitric oxide in endotoxemia and hemorrhagic shock and proposed therapeutic strategies for
manipulation of nitric oxide production.
Endotoxemia
Hemorrhagic shock
Effects of NO
Beneficial
by eNOS
-maintains perfusion; -maintains perfusion;
cytoprotective
iNOS
Beneficial
cytoprotective
Beneficial and toxic- Beneficial and toxicdepending on site
can induce tissue damage and
of production and
promote inflammation with
microenvironment
sustained shock
Therapeutic strategy
Inhibition of eNOS
Inhibition of iNOS
NO scavengers
NO donors
Avoid
Avoid
Possibly desirable-
Probably desirable-
to reduce to limit
exaggerated inflammatory
cytotoxicity and
response and development of
combat hypotension
MODS
Probably desirable-quench
extracellular NO without
inhibition of eNOS or iNOS;
supplement antioxidant
defenses
Possibly desirable-sitespecific donors
without adverse systemic
side effects;
limited availability
Probably desirable-quench
extracellular NO without
inhibition of eNOS or iNOS;
supplement antioxidant
defenses
Possibly desirable-sitespecific donors
without adverse systemic
side effects;
limited availability
Therapeutic Outlook
LINCS: L-NAME (a NO synthase inhibitor)
Patients were randomized to supportive care alone (n=15, control group) or to supportive care in
addition to L-NAME (1 mg/Kg bolus and 1 mg/Kg/h continuous IV drip for 5 h n=15). Death at one month
was 27% in the L-NAME group vs. 67% in the control group (p=0.008). Time on IABP and time on
mechanical ventilation were significantly shorter in the L-NAME group. The results of this study indicate
that NO synthase inhibitors are beneficial in the treatment of patients with refractory cardiogenic shock.
Inducible Nitric Oxide Synthase Inhibitors
Inducible nitric oxide synthase (iNOS)-dependent production of nitric oxide (NO) plays an important role
in inflammation. The e€ects of various naturally occurring furanocoumarins on NO production in
lipopolysaccharide (LPS)-activated RAW 264.7 macrophage cells were evaluated in vitro. The results
showed that angelicin, pimpinellin, sphondin, byakangelicol, oxypeucedanin, oxypeucedanin hydrate,
xanthotoxin, and cnidilin are potential NO production inhibitors, and their IC50 values for inhibition of
nitrite production were 19.5, 15.6, 9.8, 16.9, 16.8, 15.8, 16.6, and 17.7 mg/mL, respectively. Distinct
structure activity relationships were also revealed for the NO production inhibitory activities of these
furanocoumarins. Activities of the angelicin type such as pimpinellin and sphondin were more potent
than those of the psoralen type. Presence of a methoxy at the C6 position in the angelicin type seemed
to be essential to augment the activity. Western blot analysis demonstrated that only sphondin dosedependently inhibited the expression of the iNOS protein at 2.5±20 mg/mL. However, iNOS enzyme
activity was stimulated with LPS for 12 h and sphondin was administered (20 mg/mL) for 24 h, which did
not reasonably inhibit iNOS enzyme activity. l-NAME (100 mM), a known specific inhibitor of iNOS, was
employed as a positive control with the same protocol and showed more than 50% inhibition activity.
The results demonstrate that the NO production inhibitory activity of sphondin is due to the effect of
iNOS expression, but not by direct inhibition of iNOS enzyme activity. Thus, sphondin may act as a
potent inhibitor of NO production under tissue-damaging inflammatory conditions.
S-Methylisothiourea Sulfate, A Potent And Selective Inhibitor Of Inducible Nitric Oxide Synthase
Non-isoform-selective inhibition of NO formation, however, may lead to side effects by inhibiting the
constitutive isoform of NOS and, thus, the various physiological actions of NO. S-Methylisothiourea
sulfate (SMT) is at least 10- to 30-fold more potent as an inhibitor of inducible NOS (iNOS) in
immunostimulated cultured macrophages (EC50, 6 ,AM) and vascular smooth muscle cells (EC50, 2 ,uM)
than NG-methyl-L-arginine (MeArg) or any other NOS inhibitor yet known. The effect of SMT on iNOS
activity can be reversed by excess L-arginine in a concentration-dependent manner.
Enhanced formation of NO following the induction of iNOS contributes importantly to the
circulatory failure (hypotension and vascular hyporeactivity to vasoconstrictor agents) in
circulatory shock of various etiologies.
SMT dose-dependently reverses (0.01-3 mg/kg) the hypotension and the vascular
hyporeactivity to vasoconstrictor agents caused by endotoxin [bacterial lipopolysaccharide]
SMT, a potent and selective inhibitor of iNOS, may have considerable value in the therapy of
circulatory shock of various etiologies and other pathophysiological conditions associated with
induction of iNOS.
SMT, or other iNOS-selective inhibitors, are likely to have fewer side effects which are related to
the inhibition of eNOS, such as excessive vasoconstriction and organ ischemia), increased
platelet and neutrophil adhesion and accumulation, and microvascular leakage.
Iron Chelates Bind Nitric Oxide
Nitric oxide (NO), a short-lived potent vasodilator, was first described as the endothelium-derived
relaxation factor (EDRF). The formation of NO from the guanidine nitrogen group of L-arginine is
catalyzed by a group of enzymes termed constitutive (cNOs) and inducible (iNOs) NO synthases. The
inducible form is not present constitutively in mammalian cells but is induced by proinflammatory
stimuli such as bacterial lipopolysaccharide (LPS), Corynebacterium parvum, and the cytokines tumor
necrosis factor-a, interleukin-1, or interferon-y, individually or in combination. Excess production of NO
is reported to be associated with the development of hypotension associated with endotoxemia and
sepsis.
Electrochemical studies show that FeIII-(DTPA)2- binds NO stoichiometrically upon reduction to iron(II)
at biologically relevant potentials to form a stable NO adduct. In contrast, FeI"I(HDFB)+ is a stable and
efficient electrocatalyst for the reduction of NO to N20 at biologically relevant potentials. These results
suggest that the mechanism of protection against death by septic shock involves NO scavenging and that
particularly effective drugs that operate a low dosages may be designed based on the principle of redox
catalysis. These complexes constitute a new family of drugs that rely on the special ability of transition
metals to activate small molecules.
Iron complexes could act as general NO scavengers and provide protection against septic shock. Iron
complexes are capable of forming relatively stable NO adducts. Metal complexes, and in particular iron
chelators, could act as "molecular sponges," mopping up the excess NO produced during septic shock.
Iron chelators can sequester and (as for 2) catalyze conversion of NO to benign products. Demonstration
of mechanistic aspects of septic shock protection in vivo, including interaction with other free radicals,
may be hampered by the detection limits of current analytical techniques. To detect the NO Fe-DETC
complexformation in livers of LPS-treated mice by the electron paramagnetic resonance,
After screening a library of metal chelators and chelates [Fe(III)(H2DTPA)] and [Fe(III)(HDFB)]+ offered
the highest mortality decrease in an experimental model of septic shock. The Fe(II) form of both
complexes can bind NO, which appears to be related to their biological function.
Survival was greatly enhanced by the administration of 4 or 2 either 2 h before and at the time of or 30
min after LPS. In contrast, the Fe3+-free ligands of these compounds, 3 and 1, were less protective when
administered before and at the time of LPS and virtually ineffective when administered after LPS. The
clear advantage of 4 over 2 when administered after LPS was observed over a large number of
experiments [76% survival with 4 (n = 102 mice) and 38% survival with 2 (n =64 mice)].
The hydroxamic acid siderophore ferrioxamine B [Fe"'(HDFB)+] and the iron complex of diethylenetriamine-pentaacetic acid [FeI"(DTPA)2i] protected mice against death by septic shock induced by
Corynebacterium parvum + lipopolysaccharide. Although Fem(DTPA)2- was somewhat more effective
than FeI"(HDFB)+, the iron-free ligand H4DFB+ was significantly more effective than DTPA. The
hydroxamic acid chelator has a much higher iron affinity than the amine carboxylate, allowing for more
efficient formation of the FeI"(HDFB)+ complex upon administration of the iron-free ligand.
Efficacy of Treatment With the Iron (III) Complex of Diethylenetriamine Pentaacetic Acid
Bacteremia and septic shock also are associated with overproduction of free radicals such as hydroxyl,
superoxide, and carbon- and oxygen-centered radicals. In addition, nitric oxide (NO) overproduction is at
least partly responsible for the vasodilation that causes a reduction in mean systemic arterial pressure
(MSAP) and organ perfusion pressure during septic shock. This overproduction of NO likely results from
early activation of the endothelial constitutive form of NO synthase followed by induction of the
inducible form of NO synthase via TNF and IL-1.
The simultaneous increase and further reaction of NO with superoxide, which yields the oxidant
peroxynitrite anion, occurs in cellular systems in response to inflammatory mediators. In addition, in
sepsis-associated adult respiratory distress syndrome (ARDS), the presence of nitrotyrosine residues
(formed by reaction of peroxynitrite and the tyrosine residues of proteins) are apparent throughout the
lung.
administration of the iron (III) complex of diethylenetriamine pentaacetic acid (DTPA iron (III), prevented
death in Corynebacterium parvum 1 LPS-treated mice. Using electrochemistry, the binding of NO to
DTPA iron (II) is confirmed. The DTPA iron (II) form can be easily formed by common biological
reductants, because the potential for the iron (III/II) couple is E = 0.22.
Treatment with DTPA iron (III) resulted in a significant decrease in mortality compared to the untreated
controls. The efficacy of DTPA iron (III) increased when given to mice 2 h or more after infection. The
best results were observed when DTPA iron (III) was given 5 h after infection.
The iron (III) complex of diethylenetriamine pentaacetic acid (DTPA iron [III]) protected mice and
baboons from the lethal effects of an infusion with live LD 100 Escherichia coli. In mice, optimal results
were obtained when DTPA iron (III) was administered two or more hours after infection. Prevention of
death occurred in spite of the fact that the adverse effects of TNFa were well underway in the mouse
model.
In septic baboons, survival was observed after administration of two doses of DTPA iron (III) at 2.125
mg/kg, the first one given before, or as late as 2 h after, severe hypotension. Administration of DTPA
iron (III) did not alter mean systemic arterial pressure, but did protect baboons in the presence of high
levels of TNFa and free radical overproduction. Furthermore, exaggerated production of nitric oxide was
attenuated. Because of its ability to interact in vitro with free radicals, its poor cell permeability, and its
short half-life, we postulate that DTPA iron (III) and/or its reduced form may have protected the mice
and baboons by sequestration and subsequent elimination of free radicals (including nitric oxide) from
their systems. (J. Clin. Invest.1996. 98:192–198.)
Inhibitor Of Poly(Adenosine 5'-Diphosphate-Ribose) Synthetase
Poly(adenosine 5'-diphosphate [ADP]-ribose) synthetase (PARS) is a nuclear enzyme which, when
activated by DNA singlestrand breaks, initiates an energy-consuming, inefficient metabolic cycle by
transferring ADP-ribose units to nuclear proteins. The result of this process is a rapid depletion of
intracellular oxidized nicotinamide adenine dinucleotide and adenosine 5'-triphosphate energetic pools,
which slows the rate of glycolysis and mitochondrial respiration, leading to cellular dysfunction and
death.
Reactive oxygen-centered radicals (superoxide, hydroxyl radicals, singlet oxygen, and hydrogen
peroxide) and peroxynitrite (a reactive oxidant produced from the reaction of superoxide and nitric
oxide) are powerful triggers of DNA single strand breakage, and they induce activation of a cell suicide
cycle governed by PARS in various cell types in vitro.
Multiple reports implicated a role of PARS activation in the pathophysiology of endotoxic shock,
hemorrhagic shock, and various forms of ischemia-reperfusion injury.
Twenty pigs were chronically instrumented with intracardiac transducers to measure left ventricular
pressure, sonomicrometer crystals in the left ventricle to measure short axis diameter, an ultrasonic
flow meter to measure cardiac output, and catheters in the pulmonary artery and aorta to measure
blood pressures and collect samples. By using a randomized study design, either the novel potent PARS
inhibitor PJ34 (10 mg/kg for 1 hr, 2 mg·kg 1·hr 1 for 96 hrs) or placebo to pigs immediately before
intraperitoneal implantation of Escherichia coli 0111.B4 (2.3 0.1 1010 colony-forming units/kg)-laden
fibrin clots to produce peritonitis and bacteremia.
PJ34 treatment significantly attenuated this cytokine response. The formation of peroxynitrite and the
activation of PARS were confirmed in hearts and lungs of the septic pigs by the immunohistochemical
detection of nitrotyrosine and poly(ADP-ribose), respectively. Inhibition of PARS with PJ34 abolished
poly(ADP-ribose) formation in septic animals.
Cardiac inotropicity was evaluated by analysis of percentage of short axis diameter shortening (onedimensional ejection fraction). Bacteremia induced a rapid and progressive loss of inotropy until death
in vehicle-treated pigs. A similar decline was observed in the first 6 hrs in PJ34 pigs. This decline was
reversed on all subsequent days. Control pigs exhibited rapid and significant increases in systemic
vascular (SVR) and pulmonary vascular (PVR) resistances.
This experimental model mimics many aspects of the human sepsis syndrome. Therefore, the positive
survival benefit of PARS inhibition suggests a potential utility of PARS inhibitors in human sepsis
management. PARS activation is triggered by DNA single-strand breakage.
The current work, demonstrating increased poly(ADP-ribose) staining in the heart of septic pigs, may
point toward the importance of a myocardial, PARS dependent cardiodepressive mechanism in the
current model of shock. This hypothesis is supported by the following findings:
a) free radicals cause myocardial dysfunction and injury in a PARS dependent fashion in vitro;
b) in the current study, pharmacologic inhibition of PARS with PJ34 markedly improved myocardial
function; and
c) in prior studies, pharmacologic inhibition of PARS markedly improved myocardial contractile
function in hypoxic-reoxygenated hearts as well as in a porcine model of hemorrhagic shock.
Treatment with a potent PARS inhibitor improved survival and cardiovascular status and attenuated an
important mediator component of the inflammatory response in a lethal porcine model of sepsis. (Crit
Care Med 2002; 30:974 –980).
Decrease of the inflammatory response and induction of the Akt/protein kinase B pathway by poly(ADP-ribose) polymerase 1 inhibitor
The lack of efficacy of anti-inflammatory drugs, anti-coagulants, anti-oxidants, etc. in critically ill patients
has shifted interest towards developing alternative treatments. Since inhibitors of the nuclear enzyme
poly-(ADP-ribose) polymerase (PARP) were found to be beneficial in many pathophysiological conditions
associated with oxidative stress and PARP-1 knock-out mice proved to be resistant to bacterial
lipopolysaccharide (LPS)-induced septic shock, PARP. The mechanism of the protective effect of a
potent PARP-1 inhibitor, PJ34 was studied in LPS-induced (20 mg/kg, i.p.) septic shock in mice. We
demonstrated a significant inflammatory response by magnetic resonance imaging in the dorsal
subcutaneous region, in the abdominal regions around the kidneys and in the inter-intestinal cavities.
We have found necrotic and apoptotic histological changes as well as obstructed blood vessels in the
liver and small intestine. Additionally, we have detected elevated tumor necrosis factor-a levels in the
serum and nuclear factor kappa B activation in liver of LPS-treated mice. Pre-treating the animals with
PJ34 (10 mg/kg, i.p.), before the LPS challenge, besides rescuing the animals from LPS-induced death,
attenuated all these changes presumably by activating the phosphatidylinositol 3-kinase–Akt/protein
kinase B cytoprotective pathway.
PJ34, a novel, potent PARP-1 inhibitor was found to protect against LPS induced tissue damage. PARP
inhibitors protected Langendorff-perfused hearts against ischemia-reperfusion induced damages by
activating the PI3-kinase–Akt pathway. The importance of the PI3-kinase–Akt pathway in LPS induced
inflammatory mechanisms has gained support, raising the question whether this pathway was involved
in the effect of PJ34 on LPS-induced septic shock.
Among all the observed LPS-induced inflammatory responses, we found the most characteristic and
most pronounced increases in the gastro-intestinal tract, but no signal increase could be observed inside
the kidneys and in skeletal muscle, in the paravertebral or in the femoral muscle. All increases in signal
intensities were significantly attenuated in mice treated with PJ34.
Effect of PJ34 on survival of LPS-treated mice
PARP-1 inhibitor significantly protected the animals against LPS-induced death, with 86 and 43%
surviving mice, respectively.
PJ34 treatment itself did not induce death or any obvious damage.
Effect of PJ34 on LPS-induced NF-kB activation
LPS treatment in the lung caused a significant increase in NF-kB activation that was slightly but
not statistically significantly attenuated by PJ34 pre-treatment.
in contrast to the lung, NF-kB activation in the liver was prevented by PJ34 pre-treatment
The other tissue with observable LPS-induced pathological changes was the small intestine. Atrophy of
villi may reflect the diarrhea observed in the LPS-treated animals and is in agreement with the results of
Abreu et al. who found a Fas-mediated apoptosis in intestinal epithelial cells that was sensitised by
inhibitors of PI3-kinase and opposed by expressing constitutively active Akt.
Pre-treatment of the animals with a novel, potent PARP-1 inhibitor, PJ34, diminished the thoracic and
abdominal inflammatory responses as revealed by T2 imaging, and abolished the above mentioned
pathological changes.
The protective role of PARP inhibitors in septic shock is likely to be more complex than merely
the regulation of NF-kB/Rel-dependent gene expression.
Activation of the PI3-kinase–Akt/protein kinase B cytoprotective pathway is likely to contribute
to the protective effects of PARP inhibitors in shock and inflammation.
Carboxy-PTIO On Hemodynamic And Blood Gas Changes
Infusion of LPS caused a marked decrease in mean arterial pressure (MAP), metabolic acidosis,
and hypoxia. These effects were reversed by co-administration of carboxy-PTIO, without
affecting other hemodynamic parameters. In control animals, neither hemodynamic nor blood
gas parameters changed with or without carboxy-PTIO.
These results indicate that carboxy-PTIO attenuates LPS-induced hypotension, metabolic
acidosis, and hypoxia by scavenging excess NO from the circulation without affecting NO
synthase (NOS) activity. An NO scavenger, carboxy-PTIO, may be preferable to non-selective
NOS inhibitors for the treatment of human septic shock.
Asymmetrical Dimethyl Arginine Levels
Overwhelming infection with resultant multiple organ failure, which has been termed the 'sepsis
syndrome' , is a devastating illness with an incidence of 3 per 1,000 population per annum. It has been
characterised as a dysregulation of inflammation in response to infection attributable to a combination
of excessive inflammation, disseminated coagulopathy and disruption of the integrity of microvascular
endothelium.
Asymmetrical dimethyl arginine (ADMA) is an endogenous non-selective inhibitor of nitric oxide
synthase that may influence the severity of organ failure and the occurrence of shock secondary
to an infectious insult. Levels may be genetically determined by a promoter polymorphism in a
regulatory gene encoding dimethylarginine dimethylaminohydrolase II (DDAH II)
A prospective observational study was designed, and 47 intensive care unit (ICU) patients with
severe sepsis and 10 healthy controls were enrolled. Serum ADMA and IL-6 were assayed on
admission to the ICU and seven days later. Allelic variation for a polymorphism at position -449
in the DDAH II gene was assessed in each patient.
ADMA levels and Sequential Organ Failure Assessment scores were directly associated on day
one (p = 0.0001) and day seven (p = 0.002). The degree of acidaemia and lactaemia was directly
correlated with ADMA levels at both time points (p < 0.01). On day seven, IL-6 was directly
correlated with ADMA levels (p = 0.006). The variant allele with G at position -449 in the DDAH II
gene was associated with increased ADMA concentrations at both time points (p < 0.05).
Severity of organ failure, inflammation and presence of early shock in severe sepsis are
associated with increased ADMA levels. ADMA concentrations may be influenced by a
polymorphism in the DDAH II gene.
Several studies have added to the confusion surrounding the role of NO by demonstrating no effect of
NO or NOS inhibition on the myocardium or on b-adrenergic responsiveness. Nevertheless, in most
studies, low-to-moderate doses of iNOS inhibitors restore myocardial contractility in hearts exposed to
proinflammatory cytokines, whereas at higher doses, the effects are reversed. This finding may indicate
that small amounts of NO produced by iNOS may be necessary to maintain contractility and can be
cardio-protective in experimental sepsis.
References
VM Victor, K J McCreath and M Rochaa. Recent Progress in Pharmacological Research of Antioxidants in
Pathological Conditions: Cardiovascular Health. Recent Patents on Anti-Infective Drug Discovery, 2006,
1, 17-31 17.
G Cottera, E Kaluskia, O Miloa, A Blatta, et al. LINCS: L-NAME (a NO synthase inhibitor) In the treatment
of refractory Cardiogenic Shock. A prospective randomized study. The European Society of Cardiology.
2012. doi:10.1016/S0195-668X(03)00193-3 http://eurheartj.oxfordjournals.org/
Ve Laubach, Eg Shesely, O Smithies, Pa Sherman. Mice lacking inducible nitric oxide synthase are not
resistant to lipopolysaccharide-induced death. Proc. Natl. Acad. Sci. USA 1995; 92:10688-10692,
Genetics
NS Shah and TR Billiar. Role of Nitric Oxide in Inflammation and Tissue Injury during Endotoxemia and
Hemorrhagic Shock. Environ Health Perspect 106(Suppl 5):1139-1143 (1998).
http://ehpnetl.niehs.nih.gov/docs/1998/Suppl-5/1139-1 143shah/abstract.html
CC Wang, JE Lai, LG Chen, KY Yen, et al. Inducible Nitric Oxide Synthase Inhibitors of Chinese Herbs.
Part 2: Naturally Occurring Furanocoumarins’. Bioorganic & Medicinal Chemistry 2000; 8:2701-2707.
MJ O'Dwyer, F Dempsey, V Crowley, DP Kelleher, R McManus, T Ryan. Septic shock is correlated with
asymmetrical dimethyl arginine levels, which may be influenced by a polymorphism in the
dimethylarginine dimethylaminohydrolase II gene: a prospective observational study. Critical Care 2006;
10:R139 (doi:10.1186/cc5053) http://ccforum.com/content/10/5/R139
C Szab, Gj Southan, And C Thiemermann. Beneficial effects and improved survival in rodent models of
septic shock with S-methylisothiourea sulfate, a potent and selective inhibitor of inducible nitric oxide
synthase. Proc. Natl. Acad. Sci. USA 1994; 91:12472-12476. Pharmacology
Wm Kazmierski, G Wolberg, Jgwilson, et al. Iron chelates bind nitric oxide and decrease mortality in an
experimental model of septic shock. Proc. Natl. Acad. Sci. USA 1996;93:9138-9141.
L Molina, S Studenberg, G Wolberg, W Kazmierski, et al. Efficacy of Treatment With the Iron (III)
Complex of Diethylenetriamine Pentaacetic Acid in Mice and Primates Inoculated With Live Lethal Dose
100 Escherichia coli. J. Clin. Invest 1996; 98(1): 192-198. 0021-9738/96/07/192/07
N Kayhan, B Funke, LO Conzelmann, H Winkler, et al. The adenosine deaminase inhibitor erythro-9-[2hydroxyl-3-nonyl]-adenine decreases intestinal permeability and protects against experimental sepsis: a
prospective, randomised laboratory investigation. Critical Care 2008, 12:R125 (doi:10.1186/cc7033)
http://ccforum.com/content/12/5/R125
RD Goldfarb, A Marton, É Szabó, L Virág, et al.. Protective effect of a novel, potent inhibitor of
poly(adenosine 5'-diphosphate-ribose) synthetase in a porcine model of severe bacterial sepsis. Crit
Care Med 2002; 30:974 –980.
B Veres, F Gallyas Jr, G Varbiro, Z Berente, et al. Decrease of the inflammatory response and induction of
the Akt/protein kinase B pathway by poly-(ADP-ribose) polymerase 1 inhibitor in endotoxin-induced
septic shock. Biochemical Pharmacology 2003; 65: 1373–1382.
C Martinez, C Abad, M Delgado, A Arranz, et al. Anti-inflammatory role in septic shock of pituitary
adenylate cyclase-activating polypeptide receptor. PNAS 2002; 99(2):1053–1058. doi 10.1073
pnas.012367999
endogenously produced VIP and PACAP are participants of the natural anti-inflammatory
machinery.
VIP and PACAP are two attractive candidates for the development of therapies against acute
and chronic inflammatory diseases, septic shock, and autoimmune diseases