Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
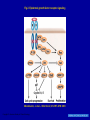
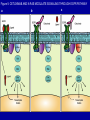
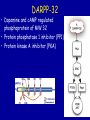
Targeting the Targets: the EGFR and HER2 stories Dr. Mark Basik Potential Conflict of Interest • Research Grant / 2006-2008 – Chemokine Therapeutics TARGETING THE TARGETS: THE EGFR AND HER2 STORIES Mark Basik MDCM Segal Cancer Center McGill University OUTLINE • EGFR and colorectal cancer • HER2 and breast cancer The HER family of receptors Fig 1. Mechanisms of receptor activation Mendelsohn, J. et al. J Clin Oncol; 21:2787-2799 2003 Copyright © American Society of Clinical Oncology Fig 2. Epidermal growth factor receptor signaling Mendelsohn, J. et al. J Clin Oncol; 21:2787-2799 2003 Copyright © American Society of Clinical Oncology EGFR as target • 65-70% colorectal cancers express EGFR (IHC) • Correlation with poor prognosis and more aggressive disease Dakocytomation EGFR as target Anti-EGFR antibodies • Cetuximab (Imclone/BMS): chimeric molecule: mouse MoAb vs ligand-binding site of EGFR+human IgG constant region fragment – Approved FDA 2004 for irinotecan-resistant metastatic colorectal cancer • Panitumumab (Amgen) :fully human IgG2 MoAb – Approved FDA 2006 for resistant EGFR-expressing metastatic colorectal cancer EGFR expression not required for response Chung et al, JCO 2005 Phase II Pharmacogenomic exploratory trial • 111 patients with metastatic colorectal cancer were enrolled in a trial of cetuximab monotherapy. • Transcriptional profiling was conducted on RNA from pre-treatment metastatic site biopsies from all patients to identify genes whose expression correlates with best clinical responses. • EGFR, B-RAF and K-RAS mutation analyses and EGFR gene copy number analyses were performed on DNA from pre-treatment biopsies. Affymetrix mRNA level Figure 2: mRNA LEVELS OF EGFR LIGANDS EPIREGULIN AND AMPHIREGULIN 3500 Epiregulin 3000 CR/PR SD non-responders 2500 2000 1500 1000 Affymetrix mRNA level 500 0 7000 Amphiregulin 6000 5000 4000 3000 2000 1000 0 Subjects Codons 12, 13 in exon 1 FFPE tissue samples PLoS Med. 2005 KRAS mutations and colorectal cancer • Activating missense K-Ras mutations – 40% colon cancers, 95% pancreatic cancers • Approximately 90% of the activating mutations are found in codons 12 (wild-type GGT) and 13 (wild-type GGC) of exon 1 and ~5% in codon 61 (wild-type CAA) located in exon 2 (8– 10). • Previous studies from various countries have revealed specific point mutations in codons 12 and 13. The most frequently observed types of mutations are G>A transitions and G>T transversions Figure 4: K-Ras MUTATION ANALYSIS 3 27 26 24 Disease Control Group (11%) Non-responders (51%) Mutant at K-Ras codon 12 or 13 Wild Type b high low Patients Free of Tumor Progression (%) EREG AREG high low (days) c (days) Patients Free of Tumor Progression (%) a Patients Free of Tumor Progression (%) Figure 3: PROGRESSION-FREE SURVIVAL CURVES K-RAS mutant wild type Khambata-Ford et al, JCO 2007 Time (days) Figure 5: CETUXIMAB AND K-RAS MODULATE SIGNALING THROUGH EGFR PATHWAY a b c CLINICAL VALIDATION • Predictive vs prognostic biomarkers – Is the marker indicating merely bad disease – How much of the drug effect is real? • A randomized clinical trial can definitely establish predictive value of biomarker Original Article K-ras Mutations and Benefit from Cetuximab in Advanced Colorectal Cancer Christos S. Karapetis, M.D., Shirin Khambata-Ford, Ph.D., Derek J. Jonker, M.D., Chris J. O'Callaghan, Ph.D., Dongsheng Tu, Ph.D., Niall C. Tebbutt, Ph.D., R. John Simes, M.D., Haji Chalchal, M.D., Jeremy D. Shapiro, M.D., Sonia Robitaille, M.Sc., Timothy J. Price, M.D., Lois Shepherd, M.D.C.M., Heather-Jane Au, M.D., Christiane Langer, M.D., Malcolm J. Moore, M.D., and John R. Zalcberg, M.D., Ph.D. N Engl J Med Volume 359(17):1757-1765 October 23, 2008 Kaplan-Meier Curves for Overall Survival According to Treatment Karapetis CS et al. N Engl J Med 2008;359:1757-1765 Kaplan-Meier Curves for Overall Survival According to K-ras-Mutation Status among Patients Receiving Supportive Care Alone Karapetis CS et al. N Engl J Med 2008;359:1757-1765 Conclusion • Patients with a colorectal tumor bearing mutated K-ras did not benefit from cetuximab, whereas patients with a tumor bearing wild-type K-ras did benefit from cetuximab • The mutation status of the K-ras gene had no influence on survival among patients treated with best supportive care alone BRAF mutations • Exclusive of KRAS mutations in CRC • 13% of CRCs Fig 1. KRAS and BRAF mutations correlate with lack of response to treatment with monoclonal antibodies targeting epidermal growth factor receptor Di Nicolantonio, F. et al. J Clin Oncol; 26:5705-5712 2008 Copyright © American Society of Clinical Oncology Figure 1. Kaplan-Meier cumulative PFS on the basis of PIK3CA and KRAS mutational status and PTEN protein expression in mCRC patients treated with panitumumab and cetuximab Sartore-Bianchi, A. et al. Cancer Res 2009;69:1851-1857 Copyright ©2009 American Association for Cancer Research WHY KRAS? KRAS mutation detection • FFPE: use enough template DNA • Direct sequencing = gold standard (min 20-50% mutant/WT ratio) • HRMA to be followed by sequencing February 2nd, 2009 Anti-EGFR TKIs (lung cancer) INTRINSIC RESISTANCE TO CETUXIMAB • MUTATIONS & MUTATIONS • LOW AUTOCRINE SIGNALING Acquired resistance to cetuximab • Cell line model (NSCLC) 6 months of cetuximab • Increased steady-state EGFR expression due to altered traffic and degradation • Activation of HER2, HER3 and cMET • No clinical trial material for acquired resistance Wheeler DL et al. Oncogene 2008 ErbB-2/Her-2/neu amplification in breast cancer 17 • 20-30% Amplification • 17q12 • Associated with: 1) Adverse prognosis 2) Aggressive tumors • Effect: cell proliferation, survival, angiogenesis, migration ErbB-2 TRASTUZUMAB (HERCEPTIN) • Humanized anti-ErbB-2 monoclonal antibody • Recognizes extracellular portion of ErbB-2 • Mechanisms of Action – – – – – – – ???? Receptor endocytosis and degradation Decreased AKT signaling (PTEN activation) ADCC Induction of apoptosis Cell cycle arrest Inhibition angiogenesis Trastuzumab response rate • Monotherapy (metastatic breast cancer): 12-34% response • Median duration 9 months • Most patients with initial response acquire resistance after 1 year Mechanisms of intrinsic resistance 1. Mechanisms involving ErbB-2: • • • • Direct implication of ErbB-2 function or localization Inhibition of trastuzumab binding to ErbB-2 (e.g. MUC4) Increased circulating ECD-HER2 (not clinically validated) Increased expression of p95 form of HER2 (partially clinically validated) Mechanisms of intrinsic resistance 2. Mechanisms involving altered downstream or compensatory signaling pathways • • • • • Downregulation of p27 Loss of PTEN (expression/mutation) (partially clinically validated) Activation of IGF-1R Upregulation of TGF-alpha Increase in heat shock protein function siRNA screen: PIK3CA/PTEN Berns, Cancer Cell 2007 INTRINSIC vs ACQUIRED • Is it the same mechanism? • The issue of cell number? – Few resistant cells: acquired – Many resistant cells: intrinsic • More difficult to study acquired Resistance both in vitro and clinically: • No clinical material Chan CT et al, Breast Cancer Res Treatment, 2005 AKT activated in Herceptinresistant cells BT 0.2 mM Hcptn 1 mM Hcptn 24 hrs __ __ + __ 0.2J __ __ + __ + __ 1E __ __ __ + pAkt Akt a-tubulin Acquired resistance to herceptin: in vivo Ritter et al, CCR 2007 DARPP-32 expression in trastuzumab-resistant cells 0.2J* 0.2J BT 0.2 mM Hcptn 1 mM Hcptn __ __ + __ 1E* 1E __ __ + + __ __ __ + __ __ __ __ + + DARPP-32 a-tubulin DARPP-32 • Dopamine and cAMP regulated phosphoprotein of MW 32 • Protein phosphatase 1 inhibitor (PP1) • Protein kinase A inhibitor (PKA) T-Darpp is required and sufficient for resistance to trastuzumab in BT-474 cells Hamel S et al. Breast Cancer Res Treat 2009 DARPP-32 activates AKT Hamel S et al. Breast Cancer Res Treat 2009 DARPP-32 expression in primary breast cancers DARPP-32: anti-apoptotic effect (AKT-BCL2) Belkhiri et al, Cancer Res. 2008 Hamel S et al. Breast Cancer Res Treat 2009 Other causes of acquired resistance to herceptin • Co-operating factors – CXCR4/DARPP32 – EGFR/DARPP32 • Context dependence (ER+ vs ER-) GENE ONTOLOGY Term SKBR3 UP P value gene changed BT474 1F P value gene changed Heat shock protein binding 0.00 1.00 Alcohol metabolism 0.00 0.69 Wnt receptor signaling 0.008 0.78 Lysosome/lytic vacuole 0.02 0.83 Monosaccharide metabolism 0.03 0.86 Unfolded protein binding 0.04 0.87 Metal ion transport 0.91 0.00 Cysteine protease inhibitor activity 1.00 0.00 cAMP mediated signaling 1.00 0.01 Amine receptor activity 0.61 0.02 HERCEPTIN RESISTANCE • Intrinsic: Mutation + HER pathway alterations • Extrinsic: different pathways activated • Functional overlap • AKT the common factor LAPATINIB: resistance • small molecule ErbB2 Tk inhibitor • Intrinsic: PIK3CA mutations and PI3K hyperactivation (Eichhorn et al, Cancer Res 2009) • Acquired: activation of ER signaling (Xia et al, PNAS 2006) How to overcome acquired resistance • Target the resistance factor – PI3K/mTor (e.g. NVP-BEZ235), geldanamycin • Combine with vertical or horizontal inhibitors (e.g. IGF-1R, mTOR…) • CONTINUE anti-Her therapy – – – – Lapatinib (affects MAPK > PI3K pathways) Pertuzumab Continue herceptin? HER3 as a target HER3: required for AKT-P Hsieh and Moasser, Br J Cancer 2007 Continuing Herceptin: double clinical response Von Minckwitz et al, Journal Clinical Oncology March 2009 Continuing anti-HER2 therapy? Fig 4. (A and B) The colorectal cancer cell line DiFi was transduced with either an empty vector or a BRAF V600E-encoding lentiviral vector Di Nicolantonio, F. et al. J Clin Oncol; 26:5705-5712 2008 Copyright © American Society of Clinical Oncology Tumor heterogeneity: an essential component of acquired resistance? Moroni, Lancet Oncology 2005 The analysis of tumor subclones MOLECULAR PROFILING: The cancer stem cell? THANK YOU • • • • • • A. Bouchard S. Hamel C. Ferrario A. Aguilar-Mahecha D.Mauro S. Khambata-Ford