Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
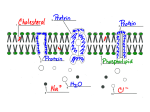
Mechanisms of Toxicity To understand how a toxicant enters an organism how it interacts with target molecules how the organism deal with the insult To provide a rational basis for interpreting descriptive toxicity data estimating the probability that a chemical will cause harmful effects establishing procedures to prevent or antagonize the toxic effects designing drugs and industrial chemicals that are less hazardous developing pesticides that are more selectively toxic for their target organisms Example for better understanding of fundamental physiologic and biochemical process Cancer and carcinogen Parkinson’s disease and MPTP Step 1-Delivery:from the site of exposure to the target Step 2a-Reaction of the ultimate toxicant with the target molecule Step 2b- Alteration of biological environment Step 3-Cellular dysfunction, injury Step 4-Inappropriate repair or adaptation Ultimate toxicant is the chemical species that reacts with the endogenous target molecule or critically alter the biological environment, initiating structural and /or functional alteration that result in toxicity. Parent compounds Metabolites of parent compounds Reactive oxygen or nitrogen species Endogenous molecules Absorption vs. presystemic elimination Influencing factors for absorption concentration of the chemical at the absorbing surface the area of the exposed site the characteristics of the epithelial layer the intensity of the subepithelial microcirculation physicochemical properties of the toxicant-lipid solubility Presystemic elimination Usually for chemicals absorbed from GI tract first pass through GI mucosal cells, liver, and lung Mechanisms facilitating distribution to a target Porosity of the capillary endothelium in the hepatic sinusoids in the renal peritubular capillaries Specialized transport across the plasma membrane ion channels protein transporters endocytosis-toxicant-protein complex membrane recycling Accumulation in cell organelles (lysosomes and mitochondria) amphipathic xenobiotics with a protonable amino group and lipophilic character Reversible intracellular binding organic and inorganic cations and PAH bind /release to melanin (polyanionic aromatic polymer) Homework p48 1. Explain the mechanism of cardiac toxicity of lipophilic local anethetics ( e.g. tetracaine, bupivacaine). 2. Why amine ( e.g. amiodarone) can cause phospholipidosis? 3. Why melanin-containing cells are more sensitive to cations and polycyclic aromatics? Mechanisms opposing distribution to a target Binding to plasma protein DDT and TCDD are bound to high M.W. protein or lipoprotein Specialized barriers (for hydrophilic toxicants) blood-brain barrier reproductive cells Distribution to storage sites (where they do not exert effects) Association with intracellular binding proteins metallothionein Export from cells by ATP dependent transports multidrug-resistance protein (P-glycoprotein) in brain cappilary endothelial cell, oocyte stem cell, and tumor cell Excretion Hydrophilic, ionized chemicals Renal glomeruli-hydrostatically filter Proximal renal tubular cells-active transport Hepatocyte Nonvolatile, highly lipophilic chemicals Excretion by the mammary gland Excretion in bile in association with biliary micelles and /or phospholipid vesicles Intestinal excretion Volatile, nonreactive toxicant Pulmonary capillaries into the alveoli Reabsorption •Renal tubule diffusion-lipid solubility, ionization (pH) carriers and transporterspeptide transporter sulfate transporter (chromate & molybdate), phosphate transporter (arsenate) •Intestinal mucosa Biliary, gastric, and intestinal excretion secretion by salivary glands and exocrine pancreas lipid solubility Toxication (metabolic activation) •Formation of electrophilic Metabolites (table3-2) molecules containing an electron-deficient atom with partial or full positive charge insertion of an oxygen atom conjugated double bonds are formed Heterolytic bond cleavage, C-O •Free radials accepting an electron from reductases (fig.3.3) losing an electron and form free radical by peroxidase homolytic fission of a covalent bond (CCl4 CCl3. , HO., Fenton reaction) •Nucleophiles (relatively uncommon) HCN from amygdalin, CO •Redox-active reactants Detoxication No functional groups add a functional group (OH,COO) by cytP450 then endogenous acid (glucuronic acid, sulfuric acid) by transferase Nucleophiles Conjugation at the nucleophilic functional group (OH, SH) Electrophiles (Metal ion, etc) conjugated with the SH of glutathione specific mechanism: epoxide hydrolase-epoxidediols, arene dihydrodiols carboxylesterase DT-diaphorase alcohol dehydrogenase Free radicals O2. - -.superoxide dismutase HOOH-glutathione peroxidase, catalase peroxyl radical-glutathione, -tocopherol, ascorbic acid ONOO--selenocysteine-containing glutathione peroxidase, selenoprotein P, oxyhemoglobin, heme-containig peroxidase, albumin peroxidase-generated free radical-electron transfer from glutathione Protein toxin-extra- and intracellular protease toxins with disulfide bond are inactivated by thioredoxin Prx(SH)2 PrxS2 2HOH chlopromazine peroxidase Homework p54 Describe at least 3 ways to prevent peroxynitrite (ONOO-) buildup. When detoxication fails Toxicants may overwhelm detoxication process exhaustion of the detoxication enzymes consumption of the cosubstrates depletion of cellular antioxidants Toxicant inactivates a detoxicating enzyme ONOO-incapacitates Mn-SOD Some conjugation reactions reversed Sometimes detoxication generates potentially harmful byproducts ex. glutathione thiyl radical (GS.) glutathione disulfide (GSSG) Attributes of target molecules DNA, protein, membrane lipids, cofactor Appropriate reactivity and/or configuration Accessibility-endogenous molecules that are in the vicinity of reactive chemicals or are adjacent to sites where they are formed ex. enzyme responsible for production of reactive metabolites or the adjacent intracellular structures Critical function-not all targets for chemicals contribute to the harmful effects ex. CO for Hb but not cytP450 Types of reactions Noncovalent binding Hydrogen bond, ionic bond ex. Interaction of toxicants with receptors, ion channels, and some enzymes Covalent binding covalent adduct formation Hydrogen abstraction R-SH, RSOH Electron transfer Fe(II)Fe(III) enzymatic reactions ADP ribosylation-diphthera toxin, cholera toxin Hydrogen abstraction Dysfunction of target molecules Activation- agonist, activator Inhibition- antagonist Alteration in conformation or structure of protein- thiol group Interference with template with the function of DNA aflatoxin bind to G: GC GA . HO 8-hydroxyguanine and 8-hydroxyadenine mispairing Destruction of target molecules Cross-linking Fragmentation spontaneous degradation after chemical attack hydrolytic degradation Neoantigen formation Covalent binding altered protein evoke immune response drug-protein adduct Toxicity not initiated by reaction with target molecules 1. Chemicals that alter H ion concentrations Acids and substance biotransformed to acids protonophoric uncoupler 2. Solvents and detergents alter the lipid phase of cell membrane and destroy transmembrane solute gradients 3. Occupying a site or a space ethylene glycol form water insoluble precipitates in the renal tubules sulfomides occupy bilirubin binding sites of albumin Dysregulation of gene expression Dysregulation of transcription Promoter region of the gene Transcription factors (TFs) ligand-activated (Table 3-4) altering the regulatory region of the genes direct chemical interaction –thalidomide/GCbox methylation of cytosine Dysregulation of signal transduction Dysregulation of the synthesis, storage, or release of the extracellular signaling molecules Systemic lupus erythemathosus Induced by Procainamide Hydrolazine Inhibit DNA methylation in CD4+T lymphocyte Overexpression of protein for inflammation TCDD hypermethylation in Insulin-like growth factor-2 gene Dysregulation of signal transduction *sinaling molecules to activate TFs ( c-FOS, c-JUN, c-Myc) that control transcriptional activity of genes that influence cell cycle Altering protein phosphorylation by kinases, by phosphatases Interfering with the GTPase activity of G protein Disrupting normal protein-protein interaction Altering the synthesis or degradation of the signaling proteins Extracellulr Signaling molecules Chemically altered signal transduction with proliferative effect Chemically altered signal transduction with antiproliferative effect Dysregulation of ongoing cellular activity dysregulation of electrically excitable cells (Table 3-5) due to an alteration in the concentration of neurotransmitters receptor function intracellular signal transduction the signal terminating process dysregulation of the activity of other cells ex.liver cells possess -1 adrenergic receptors exocrine secretory cells controlled by Ach receptor Toxic alteration of cellular maintenance Impairment of internal cellular maintenance: mechanism of cell death ATP depletion (Table 3-6) Ca accumulation (Table 3-7) ROS/RNS generation . Sustained elevation of intracellular Ca2+ can result in : 1. Depletion of energy reserve mitochondria Ca2+ uptake dissipate membrane potential continuous Ca2+ uptake and export causing oxidative injury to inner membrane impair ATP synthesis ATP consumption by the Ca2+ -ATPase (eliminate the excess Ca2+ 2. Dysfunction of microfilaments dissociation of actin filaments from -actinin and fodrin (anchor proteins) membrane blebbing 3. Activation of hydrolytic enzymes calpains phospholipases Ca2+ -Mg2+ dependent endonuclease 4. Generation of ROS and RNS Mitochondrial permeability transition (MPT) Mitochondrial inner-membrane permeability caused by opening of a proteinaceous pore (megachannel) free influx into the matrix space of protons rapid and dissipation of membrane potential and cessation of ATP synthesis osmotic influx of water mitochondrial swelling apoptosis or necrosis Induction of cell death by unknown mechanisms 1. Chemicals directly damage the plasma membrane lipid solvents, detergents, venom-drived hydrolytic enzymes 2. Xenobiotics that damage the lysosomeal membrane aminoglycoside, hydrocarbons binding to a2u-globulin 3. Toxins that destroy the cytoskeleton microfilament toxins-phalloidin and cytochalasins microtubular toxins-colchicine, 2,5-hexanedione 4. Protein phosphatase inhibitor cause hyperphosphorylation mycrocystin 5. Toxins that disrupt protein synthesis--amaitin and ricin 6.Cholesterol lowing drug statin –inhibit HMG coenzyme, myotoxi DNA repair Direct repair DNA photolyase-cleavge dimerized pyrimidine O6-alkylguanine-DNA-alkyltransferase-remove minor adducts Excision repair Base excision-DNA glycosylase Nucleotide excision-ATP dependent nuclease poly(ADP-ribose) polymerase (PARP) poly(ADP-ribose) glycohydrolase Recombination (or postreplication) repair When excision repair fail to occur before DNA replication begins Cellular repair: A strategy in peripheral neurons Macrophages-remove debris and produce cytokine and growth factors Schwann cells-proliferate and transdifferentiate from myelinating operation mode into a growth-supporting mode synthesis of cell adhension molecules (N-CAM) Elaborating excellular matrix protein for base membrane construction Producing neurotrophic factors and their receptors Comigrating with the regrowing axon, physically guide and chemically lure the axon to reinnervate the target cell Tissue repair Apoptosis: an active deletion of damaged cells Proliferation: regneration of tissue Side reactions to tissue injury Apoptosis cell shrinks apoptotic bodies phagocytosed orderly process without inflammation Necrosis cell and organelles swell membrane lysis disorderly process induce inflammation Proliferation : Regeneration of tissue Replacement of lost cells by mitosis After injury, intracellular signaling turns on § Activation of protein kinase and TF § Immediately early genes-transcription factors and like secreted protein § Delayed early genes-antiapoptotic protein § Cell cycle accelerators (cyclin D) § Cell cycle decelerators (p53, p21) § Mediators of tissue repair and side reactions Replacement of the extracellular matrix Proteins, glycosamineoglycans, glycoprotein and proteoglycan glycoconjugates Matrix metalloproteinase cytokine- IEG Growth factors Side reaction to tissue injury •Inflammation Cells and mediators tissue damage resident M secreting cytokines endothelial cells and fibroblasts release mediator Alteration of the microcirculation Accumulation of inflammatory cells (leukocyte) chemoattractant selectins on the membrane of endothelial cells ligand on the surface of leukocyte adhesion ICAM on endothelial cells integrins on the membrane of leukocyte Production of ROS and NOS M and leukocytes •Altered protein synthesis: acute-phase proteins positive acute-phase proteins minimize tissue injury and facilatating repair ex. 2-macroglobulin, 1-antiprotease inhibit lysosomal protease released from the injured cell metallothionein complexes metals Negative acute-phase proteins plasma proteins-albumin, transthyretin, transferrin Cytochrome P450 Glutathione S-transferase •Generalized reaction Cytokines evoke neurohormonal responses ex. IL-1 sickness behavior ACTH release Mechanisms of adaptation Adaptation by decreasing delivery to the target -Repression of iron absorption -Induction of ferritin and metallothionein -Induction of detoxication Adaptation by decreasing the target density or responsiveness -induction of opioid tolerance Adaptation by increasing repair -induction of enzymes repairing oxidized proteins (Fig 3-23) -induction of chaperones repairing misfolded proteins heatshock response, ER stress response -induction of enzymes repairing DNA (p53) -adaptive increase in tissue repair (NF-κB) Mechanisms of adaptation Adaptation by compensating dysfunction Adaptation to hypoxia –the hypoxia response (HIF-1) Adaptation to energy depletion-energy stress response (AMPK) Adaptation by neurohormonal mechanisms Toxicity resulting from dysrepair Tissue Necrosis Fibrosis-excessive disposition of an extracellular matrix of abnormal composition Carcinogenesis Failure of DNA repair: mutation, the initiating event in carcinogenesis Failure of apoptosis:promotion of mutation and clonal growth Failue to terminate proliferation:promotion of mutation, protooncogene overexpression, and clonal growth Nongenotoxic carcinogens:promotors of mitosis and inhibitors of apoptosis Conclusions An organism has mechanisms that 1. Counteract the delivery of toxicant, such as detoxication 2. Reverse the toxic injury, such as repair mechanisms 3. Offset some dysfunctions, such as adaptive responses Toxicity is not an inevitable consequence of toxicant exposure. Toxicity develops if the toxicant exhausts or impairs the protective mechanisms and/or overrides the adaptability of biological systems. Homework 1. Describe the types of ultimate toxicant . 2. How detoxication of free radical exert? 3. What will occur following reaction of ultimate toxicant with endogenous molecule? A. at molecular level B. at cellular level 4. What are the three major processes to impair the internal cellular maintenance and cause cell death? 5. Why is the sustained rise of intracellular calcium level harmful? 6. Describe how a cell to repair proteins, lipids, and DNA? 7. Explain the heatshock response and ER stress response. 8. What are the possible outcomes when repair fails?