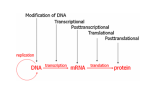
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Published OnlineFirst August 19, 2013; DOI: 10.1158/1078-0432.CCR-13-0223 Molecular Pathways: Protein Methyltransferases in Cancer Robert A. Copeland Clin Cancer Res 2013;19:6344-6350. Published OnlineFirst August 19, 2013. Updated version Cited Articles E-mail alerts Reprints and Subscriptions Permissions Access the most recent version of this article at: doi:10.1158/1078-0432.CCR-13-0223 This article cites by 40 articles, 14 of which you can access for free at: http://clincancerres.aacrjournals.org/content/19/23/6344.full.html#ref-list-1 Sign up to receive free email-alerts related to this article or journal. To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at [email protected]. To request permission to re-use all or part of this article, contact the AACR Publications Department at [email protected]. Downloaded from clincancerres.aacrjournals.org on December 3, 2013. © 2013 American Association for Cancer Research. Published OnlineFirst August 19, 2013; DOI: 10.1158/1078-0432.CCR-13-0223 Clinical Cancer Research Molecular Pathways Molecular Pathways: Protein Methyltransferases in Cancer Robert A. Copeland Abstract The protein methyltransferases (PMT) constitute a large and important class of enzymes that catalyze sitespecific methylation of lysine or arginine residues on histones and other proteins. Site-specific histone methylation is a critical component of chromatin regulation of gene transcription—a pathway that is often genetically altered in human cancers. Oncogenic alterations (e.g., mutations, chromosomal translocations, and others) of PMTs, or of associated proteins, have been found to confer unique dependencies of cancer cells on the activity of specific PMTs. Examples of potent, selective small-molecule inhibitors of specific PMTs are reviewed that have been shown to kill cancers cells bearing such oncogenic alterations, while having minimal effect on proliferation of nonaltered cells. Selective inhibitors of the PMTs, DOT1L and EZH2, have entered phase I clinical studies and additional examples of selective PMT inhibitors are likely to enter the clinic soon. The current state of efforts toward clinical testing of selective PMT inhibitors as personalized cancer therapeutics is reviewed here. Clin Cancer Res; 19(23); 6344–50. 2013 AACR. Background Posttranslational modifications of histone proteins play a critical role in defining the local structure of chromatin (the complex of DNA and histone proteins that make up chromosomes) and thereby control entire programs of gene transcription within cells (1). Both reversible small-molecule (e.g., acetylation, methylation, and phosphorylation) and protein (e.g., ubiquitination and sumoylation) modifications of histone are known to affect nucleosome compacting within chromatin and also to serve as recognition loci for binding of transcription factors, polymerases, and auxillary proteins that either facilitate or antagonize gene transcription (2). Among these various histone modifications (Fig. 1A), methylation of lysine and arginine residues seems to play a particularly important role in control of gene transcription programs (3–5). These modifications are catalyzed by a class of group-transfer enzymes known as the protein methyltransferases (PMT; refs. 3, 6). The PMT enzyme class is composed of two distinct families of enzymes, based on their active site structures and on the amino acid to which they transfer methyl groups: the protein lysine methyltransferases (PKMT) and the protein arginine methyltransferases (PRMT; ref. 6). There is one exception to this general structural bifurcation of the PMT class. The enzyme DOT1L is biochemically a lysine methyltransferase, but its active site structure is most Author's Affiliation: Epizyme, Inc., Cambridge, Massachusetts Corresponding Author: Robert A. Copeland, Epizyme, Inc., 400 Technology Square, Cambridge, MA 02139. Phone: 617-500-0707; Fax: 617-3490707; E-mail: [email protected] doi: 10.1158/1078-0432.CCR-13-0223 2013 American Association for Cancer Research. 6344 closely aligned with that of the protein arginine methyltransferases (6). On the basis of active site structure, a number of enzymes that had been previously annotated as RNA methyltransferases can also be included within the family of PRMTs, and some of these enzymes have subsequently been shown biochemically to catalyze both RNA and protein methylation (6, 7). Thus, the PMT class is composed of 96 enzymes within humans. Reaction fidelity is variable among the PMTs. In some cases, a single enzyme is responsible for site-specific methylation of a unique histone location (e.g., DOT1L methylation of H3K79; see below). In other cases, several PMTs seem to methylate a common histone site but may do so in a gene-specific manner (Fig. 1B). In yet other cases, a single enzyme may methylate multiple protein substrates. The PMTs constitute an interesting target class for cancer therapeutics both because of the critical role these enzymes play in controlling cellular programs of gene transcription and because, increasingly, bioinformatic surveillance of cancer genome databases are showing that a number of these enzymes are implicated in specific human cancers (5, 8); in a recent review, for example, Copeland listed over 10 PMTs for which genetic alterations are found in specific human cancers (8). PMTs may play critical roles in cancer cells in general, but their greatest value as therapeutic targets may be in cases where the oncogenic alterations of PMTs (or of other proteins that impact PMT activity; see below) associated with specific human cancers have been shown to confer to cancer cells a unique dependency on the enzymatic activity of the altered PMT for proliferation and/or survival (i.e., an addiction to PMT activity). Hence, a reasonable working hypothesis is that selective inhibition of the affected PMT would result in cell killing of cancer Clin Cancer Res; 19(23) December 1, 2013 Downloaded from clincancerres.aacrjournals.org on December 3, 2013. © 2013 American Association for Cancer Research. Published OnlineFirst August 19, 2013; DOI: 10.1158/1078-0432.CCR-13-0223 PMTs in Cancer A Ac R K CH3 K Ub K Figure 1. Chromatin modification affects gene transcription. A, chromatin consists of histone proteins (red) around which chromosomal DNA (blue) is wrapped. The histone proteins undergo a variety of posttranslational modifications, including methylation of arginine (R) and lysine (K) residues catalyzed respectively by the PMT families of arginine methyltrasferases (RMT) and lysine methyltransferases (KMT). These posttranslational modifications affect the state of compacting of nucleosomes (histone-DNA units) along the chromosomes. Regions of chromosomes can transition from the open, transcriptionally permissive state referred to as euchromatin to the more compact, transcriptionally respressive state known as heterochromatin. B, PMTs catalyze methylation of specific lysine and arginine residues on histones H3 and H4. The enzymes that catalyze methylation of a specific histone residue are listed in the boxes. PMTs • Arginine (RMTs) • Lysine (KMTs) S PO4 CH3 CpG Euchromatin Transcriptionally permissive Heterochromatin Transcriptionally repressive B Site-specific methylation of histones H3 & H4 CARM1 PRMT6 H3 MLL1 MLL2 MLL3 MLL4 MLL5 SET1A SET1B ASH1 SET7/9 PRMT5 EZH1 EZH2 CARM1 NDS1 NSD2 NSD3 SET2 SMYD2 DOT1L N-A R T K ... R K S T ... K ... R K ... K ... R K S ... K ... K ... -C 2 4 8 9 17 26 27 36 79 PRMT1 PRMT5 H4 SUV39H1 SUV39H2 G9a GLP SETDB1 CLL8 RIZ1 PRDM3 PRDM16 Pr-SET7/8 SUV4 20H1 SUV4-20H2 SET9 Pr-SET7 SUV4-20 NSD1 SMYD3 N-S G R G K G G K G L G K G G A K R H R K V ...... -C 1 3 5 20 © 2013 American Association for Cancer Research cells bearing the genetic alteration with minimal affect on nonaltered cells. This would be expected to provide a significant therapeutic index in vivo, with potent elimination of affected tumor cells and limited attendant safety concerns. Preclinical animal studies of potent, selective inhibitors of two PMTs, DOT1L (9) and EZH2 (ref. 10; see below) have borne out these expectations and www.aacrjournals.org have paved the way to clinical testing of PMT inhibitors in patients with cancer. Active site structure and enzymatic mechanism of methyl transfer All PMTs use S-adenosyl methionine as a universal methyl donor in much the same way that all kinases use Clin Cancer Res; 19(23) December 1, 2013 Downloaded from clincancerres.aacrjournals.org on December 3, 2013. © 2013 American Association for Cancer Research. 6345 Published OnlineFirst August 19, 2013; DOI: 10.1158/1078-0432.CCR-13-0223 Copeland PKMTs Histone H3 C NH3+ Histone HMT N H O + O CH3 + N CH3 H3C + HMT N H O Mono-methyl lysine Lysine CH3 NH HMT N H O H3C + NH2 O N H O O O Di-methyl lysine Tri-methyl lysine CH3 CH3 CH3 CH3 PRMTs CH3 S PMT + S H 2N – – CH3 NH2+ HN HN NH HN NH3+ NH3 SAM SAH N H O O Arginine N H3C HN HMT + HN HMT N H O O Mono-methyl arginine N NH HN or N H O O Symmetrical di-methyl arginine N H O O Asymmetrical di-methyl arginine © 2013 American Association for Cancer Research Figure 2. Chemical mechanism of PMT catalysis (left) and the products of lysine (top right), and arginine (bottom right) methylation. SAM, S-adenosyl methionine ATP as a universal phosphate donor (Fig. 2). The crystal structures of a number of PMTs have been solved to atomic resolution and reveal a well-organized S-adenosyl methionine–binding pocket, which is conformationally distinct between the lysine and arginine methyltransferases (3, 6, 11). In both cases, however, the S-adenosyl methionine–binding pocket is juxtaposed to a long channel into which the methyl-accepting amino acid side chain binds. This arrangement allows for close proximity and molecular orbital alignment between S-adenosyl methionine and the nitrogen of lysine or arginine for facile methyl transfer. All of these enzymes transfer the methyl group directly from S-adenosyl methionine to the acceptor amino acid, so that the enzymatic reaction requires the simultaneous binding of both substrates for transfer to occur (3). These features of the enzymatic reaction of PMTs afford multiple opportunities for inhibitor interactions with the enzymes. Indeed, small-molecule PMT inhibitors of various binding modalities have been reported: S-adenosyl methionine competitive, methyl acceptor competitive, noncompetitive, etc. (5, 8). This diversity of inhibitor-binding modalities distinguishes the PMTs from other group-transfer enzymes that have been targeted for cancer therapies (e.g., the kinases) and may allow greater flexibility in choosing inhibitors that offer the greatest chance for clinical translation (12). Role of PMTs in regulation of gene transcription How site-specific methylation of histone lysines and arginines controls gene transcription is incompletely under- 6346 Clin Cancer Res; 19(23) December 1, 2013 stood at present, but it is clear that methylation at specific histone sites affects downstream transcription in different ways. For example, methylation of histone H3 on lysines 4 (H3K4) and 79 (H3K79) has been implicated in the transcriptional activation of genes. In contrast, methylation at other locations, such as H3K9 and H3K27, are involved in repression of gene transcription (13). These opposing effects of methylation of different histone sites can play a critical role in cancer tumorigenesis when alterations in the location and amplitude of specific site methylation leads to aberrant activation of oncogene transcription and/or transcriptional repression of tumor suppressor genes. It is also becoming clear that the effect of site-specific methylation of histone sites on transcription cannot be understood in isolation, but depends on the context of other modifications of proximal histone sites (14). Finally, emerging data suggest that methylation of one histone site may enhance or antagonize methylation of other histone sites. For example, data in multiple myeloma cells suggest that elevated methylation of H3K36 antagonizes methylation of H3K27, and likewise diminution of H3K36 methylation may result in elevation of H3K27 methylation levels (15). Genetic alterations of PMT activity as drivers of cancer A variety of genetic alterations are seen in members of the PMT enzyme class associated with specific human cancers. Ongoing surveillance of cancer genome databases is continuing to reveal additional examples of amplifications and mutations within PMTs and within PMT-associated proteins that seem to be specific to Clinical Cancer Research Downloaded from clincancerres.aacrjournals.org on December 3, 2013. © 2013 American Association for Cancer Research. Published OnlineFirst August 19, 2013; DOI: 10.1158/1078-0432.CCR-13-0223 PMTs in Cancer individual cancer types. Over the past decade, several examples have emerged of alterations of PMT activity that seem to be critical drivers of genetically defined cancers (16). Some of these will be described here to exemplify the diversity of diver alterations observed to impinge on this enzyme class. Indirect chromosomal translocation leading to ectopic PMT activity. MML-rearranged leukemia is a devastating form of acute leukemia that affects patients from infancy through adulthood and is also prevalent among secondary leukemia associated with the use of etoposide (and other topoisomerase II inhibitors; ref. 17). This disease is universally associated with a chromosomal translocation (11q23) affecting the MLL gene that encodes for the PMT MLL, which normally catalyzes the methylation of the H3K4 residue (18, 19). As a result of the translocation, the MLL protein loses its enzymatic active site (hence, PMT activity) and is fused to any of a number of proteins within the AF or ENL protein families (20, 21). Gene localization is conferred by recognition elements within the N-terminal region of the MLL protein, and these are retained within the various fusion proteins. Hence, the MLL fusion proteins localize to the same gene locations as wild-type MLL, but lack the ability to catalyze histone methylation at H3K4. It has been recognized for some time that this chromosomal translocation plays a causal role in MML-rearranged leukemia, but the molecular mechanism of pathogenesis was not clear. Early attention focused on the loss of PMT activity associated with the fusion proteins that results from the translocation, but this was hard to reconcile with the residual H3K4 methylation activity of the wild-type MLL protein resulting from the second, unaffected allele. In 2005, Okada and colleagues (22) showed that another enzyme, DOT1L, binds to AF10, one of the various fusion partner proteins associated with the MLL translocation, and thereby recruits DOT1L to ectopic gene locations at which the MLL fusion proteins reside. Subsequent studies revealed that other AF and ENL fusion partners were also able to directly, or indirectly recruit DOT1L to MLL fusion–bound genes (23–25). The resulting ectopic localization of DOT1L catalyzes H3K79 methylation of genes otherwise under the control of MLL-catalyzed H3K4 methylation, including proleukemogenic genes such as HOXA9 and MEIS1 (22, 26, 27). Methylation of H3K79 is a transcriptional activation mark. Thus, the ectopic localization of DOT1L, due to the universal chromosomal translocation in MML-rearranged leukemia, results in elevated transcription of a program of leukemogenic genes that in turn drive this proliferative disease. It has been hypothesized that MML-rearranged leukemia is uniquely dependent on DOT1L enzymatic activity and that selective inhibition of DOT1L would lead to specific cell killing of MML-rearranged leukemia cells. This hypothesis was confirmed by gene silencing approaches (26) and by the use of potent and selective small-molecule inhibitors of DOT1L (see below; refs. 28–30). Direct chromosomal translocations involving PMTs. WHSC1 (also referred to as MMSET or NSD2) is a PMT that catalyzes the dimethylation of H3K36 (31, 32). www.aacrjournals.org Methylation of this histone site is associated with regions that are transcriptionally active. The t(4;14) chromosomal translocation occurs in a subset of about 15% of multiple myeloma (33, 34), and patients with this translocation represent one of the worst prognostic subgroups of multiple myeloma (35). The t(4;14) translocation results in massive overexpression of WHSC1 and of fibroblast growth factor receptor 3 (FGFR3) due to the placement of the strong immunoglobulin H (IgH) intronic Em enhancer and 30 enhancer in the promoter regions of WHSC1 and FGFR3 genes, respectively. While approximately 30% of t(4;14) patients have lost expression of FGFR3, 100% retain overexpression of WHSC1, suggesting that WHSC1, rather than FGFR3, is the primary driver of disease (35). The overexpression of WHSC1 in t(4;14) translocated cells results in significantly elevated levels of dimethylated H3K36, as would be expected from elevation of catalytic enzyme levels (15). Genetic knockdown of WHSC1 or disruption of the translocated allele in t(4;14) myeloma cells results in inhibition of cellular proliferation and of tumorigenicity. As expected, genetic knockdown of WHSC1 shows an accompanying reduction in global levels of H3K36me2 (15). Point mutations affecting PMT activity. The most wellstudied example of tumorigenic point mutations affecting PMT activity is that of mutations within the catalytic domain of EZH2 in germinal center diffuse large B-cell lymphoma and follicular lymphoma subsets of non-Hodgkin lymphoma (NHL; 36–38). EZH2 (or the closely related EZH1) is the catalytic subunit of a multiprotein complex known as PRC2 (polycomb repressive complex 2) that is responsible for mono-, di-, and trimethylation of the H3K27 site. Trimethylated H3K27 is a translational repressive mark, and hypertrimethylation of H3K27 has always been found to lead to tumorigenesis in various human cancers, presumably due to the transcriptional repression of tumor suppressor genes. Mutations at Tyr641 or Ala677 have been found to occur heterozygously in NHL patient groups (36, 37) and have been shown to be change-of-function mutations with respect to the substrate specificity of the enzyme (38). The wild-type enzyme is most active in placing the first methyl group on to H3K27 and its catalytic efficiency wanes with consecutive methylation cycles. In direct contrast, the mutations associated with NHL show the exact opposite substrate specificity; they are essentially unable to put the first methyl group on H3K27, but once that first methyl is on, the mutant enzymes are able to put the second methyl on and are extremely efficient at putting the third methyl group on. In the context of disease heterozygosity, the wild-type and mutant enzymes work in concert with one another to effect a tumorigenic hypertrimethylated H3K27 phenotype (38). Two groups have now shown that potent-, selective-, small-molecule inhibitors of EZH2 selectively kill NHL cells bearing these EZH2 mutations while having essentially no effect on the proliferation of homozygous EZH2 wild-type cells (39, 40). Clin Cancer Res; 19(23) December 1, 2013 Downloaded from clincancerres.aacrjournals.org on December 3, 2013. © 2013 American Association for Cancer Research. 6347 Published OnlineFirst August 19, 2013; DOI: 10.1158/1078-0432.CCR-13-0223 Copeland Synthetic lethal associations with PMT activity. In addition to histone methylation, gene expression is also regulated by remodeling of nucleosomes in an ATPdependent manner. Of the ATP-dependent chromatin remodelers, SWI/SNF complexes are emerging as bona fide tumor suppressors, as specific inactivating mutations in several SWI/SNF subunits are found in human cancers (41). For instance, the INI1 (also known as SMARCB1 or hSNF5) subunit is inactivated in nearly all malignant rhabdoid tumors and atypical teratoid rhabdoid tumors, aggressive cancers of young children with no effective therapy (42). Tumorigenesis in INI1-deficient malignant rhabdoid tumors can be completely suppressed by tissue specific codeletion of EZH2, suggesting an antagonistic relationship between PRC2 and SWI/SNF activities (43). Recently, Knutson and colleagues (44) have shown that potent, selective inhibitors of EZH2 are effective in eradicating INI1-deficient malignant rhabdoid tumors in cell culture and in a mouse xenograft model. Hence, although the tumorigenetic driver of malignant rhabdoid tumors occurs in a pathway distinct from any PMT, it nevertheless creates a synthetic lethal relationship that confers a unique sensitivity to selective EZH2 inhibition in this disease. Clinical–Translational Advances Preclinical in vivo activity of selective PMT inhibitors Selective inhibitors of several PMTs have been described, and compounds targeting two particular PMTs, DOT1L and EZH2, have been reported to show dramatic antitumor activity in rodent models of disease (9, 10, 40, 44). The DOT1L tool compound EPZ004777 was the first PMT inhibitor reported to show antitumor effects in a disseminated mouse model of MLL-rearranged leukemia (28). Subsequent pharmacologic optimization within this chemotype series resulted in the clinical candidate DOT1L inhibitor EPZ-5676, which was also shown to eradicate MLL-rearranged tumors (100% tumor growth inhibition) in a rat xenograft model (9, 45). Two distinct EZH2 inhibitors have been shown to display significant, dose-dependent antitumor activity in mouse xenograft models of EZH2 mutant–bearing NHL tumors (60%–100% tumor growth inhibition in various tumor models) and of INI1-deficient malignant rhabdoid tumors (100% tumor growth inhibition; refs. 10, 40, 44). In all of these studies, the compounds were well tolerated by the rodents at doses that resulted in significant tumor growth inhibition, with no acute toxicity or weight loss observed. These results provide a strong foundation for the clinical testing of DOT1L and EZH2 inhibitors in genetically defined patients with cancer and also portend the general use of PMT inhibitors as personalized cancer therapeutics. Also, in all of these studies, the investigators were able to show a correlative relationship between tumor growth inhibition and diminution of the relevant histone methyl mark in tumor tissue as well as in surrogate tissues such as skin and peripheral blood mononu- 6348 Clin Cancer Res; 19(23) December 1, 2013 clear cells. The ability to measure drug-induced loss of site-specific histone methylation in surrogate tissue by noninvasive means provides a cogent basis for pharmacodynamics assessment of target engagement in clinical trials. Clinical trials At the time of this writing, two PMT inhibitors have entered phase I human clinical trials: the DOT1L inhibitor EPZ-5676 and the EZH2 inhibitor EPZ-6438. The primary goal of both of these trials is to assess the safety of the drugs and to establish the maximum tolerated dose for each. The EPZ-5676 phase I trial is divided into two parts: a dose-escalation portion to establish maximum tolerated dose and an expansion cohort of patients treated at the maximum tolerated dose. To accelerate dose escalation, eligibility is not restricted to genetically defined patients; patients with hematologic malignancies (mainly acute myelogenous leukemia and acute lymphocytic leukemia) are eligible for this portion of the trial. Once the maximum tolerated dose has been established, the expansion cohort study will begin exclusively in patients with leukemia involving the MLL rearrangement at 11q23. In this manner, the drug may be tested as early as possible in the genetically defined patients for whom it was designed. The EZH2 inhibitor EPZ-6438 is being studied in a phase I trial of relapsed or refractory malignancies that have failed all standard therapy. Again, the primary goal here is to establish the safety and define the maximum tolerated dose of the drug. As with the EPZ-5676 trial, a secondary objective is to test EPZ-6438 as early as possible in the genetically defined patients for whom the drug was designed. Hence, once the maximum tolerated dose has been established, a phase IIa trial is planned exclusively in NHL patients bearing mutations in EZH2. No data have yet been reported for either of these trials. Nevertheless, these trials represent a true watershed in the development of PMT inhibitors as personalized cancer therapeutics. The cancer community will be looking forward with great interest toward the outcomes of these trials. It is clear that these trials represent the vanguard of a much larger array of PMT inhibitors that will be entering clinical trials in the near future for testing against genetically defined groups of patients with cancer. Disclosure of Potential Conflicts of Interest R.A. Copeland has an ownership interest (including patents) in Epizyme Inc. and is a consultant/advisory board member of Mersana. Acknowledgments The author thanks Dr. Roy M. Pollock for helpful suggestions. Received June 24, 2013; revised July 21, 2013; accepted August 7, 2013; published OnlineFirst August 19, 2013. Clinical Cancer Research Downloaded from clincancerres.aacrjournals.org on December 3, 2013. © 2013 American Association for Cancer Research. Published OnlineFirst August 19, 2013; DOI: 10.1158/1078-0432.CCR-13-0223 PMTs in Cancer References 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. 17. 18. 19. 20. 21. 22. 23. Walsh CT, Garneau-Tsodikova S, Gatto GJ Jr. Protein posttranslational modifications: the chemistry of proteome diversifications. Angew Chem Int Ed Engl 2005;44:7342–72. Allis CD, Jenuwein T, Reinberg D, Caparros M-L. Epigenetics. New York: Cold Spring Harbor Press; 2007. Copeland RA, Solomon ME, Richon VM. Protein methyltransferases as a target class for drug discovery. Nat Rev Drug Discov 2009;8:724–32. Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov 2012;11:384–400. Copeland RA, Moyer MP, Richon VM. Targeting genetic alterations in protein methyltransferases for personalized cancer therapeutics. Oncogene 2013;32:939–46. Richon VM, Johnston D, Sneeringer CJ, Jin L, Majer CR, Elliston K, et al. Chemogenetic analysis of human protein methyltransferases. Chem Biol Drug Des 2011;78:199–210. Webb KJ, Lipson RS, Al-Hadid Q, Whitelegge JP, Clarke SG. Identification of protein N-terminal methyltransferases in yeast and humans. Biochemistry 2010;49:5225–35. Copeland RA. Protein methyltransferase inhibitors as personalized cancer therapeutics. Drug Discov Today Ther Strateg 2012;9:e83-e90. Pollock RM, Daigle SR, Therkelsen CA, Basavapathruni A, Jin L, Allain CJ, et al. Preclinical characterization of a potent, selective inhibitor of the protein methyltransferase DOT1L for use in the treatment of MLLrearranged leukemia [abstract]. In: Proceedings of the 54th ASH Annual Meeting and Exposition; 2012 Dec 8–11; Atlanta, GA. Washington, DC: ASH; 2012. Abstract nr 2379. Keilhack H, Yokoi A, Knutson SK, Wigle T, Warholic N, Kawano S, et al. Preclinical characterization of E7438, a potent, selective inhibitor of protein methyltransferase EZH2 with robust antitumor activity against EZH2 mutated non-Hodgkin lymphoma xenografts in mice [abstract]. In: Proceedings of the 54th ASH Annual Meeting and Exposition; 2012 Dec 8–11; Atlanta, GA. Washington, DC: ASH; 2012. Abstract nr 3712. Cheng X, Collins RE, Zhang X. Structural and sequence motifs of protein (Histone) methylation enzymes. Annu Rev Biophys Biomol Struct 2005;34:267–94. Copeland RA. Evaluation of enzyme inhibitors in drug discovery: a guide for medicinal chemists and pharmacologists. 2nd ed. New York: Wiley; 2013. Kouzarides T. Chromatin modifications and their function. Cell 2007; 128:693–705. Lee JS, Smith E, Shilatifard A. The language of histone crosstalk. Cell 2010;142:682–5. Martinez-Garcia E, Popovic R, Min D-J, Sweet SMM, Thomas PM, Zamdborg L, et al. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood 2011;117:211–20. Decarlo D, Hadden MK. Oncoepigenomics: making histone lysine methylation count. Eur J Med Chem 2012;56:179–94. Muntean AG, Hess JL. The pathogenesis of mixed-lineage leukemia. Annu Rev Pathol 2012;7:283–301. Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD, et al. MLL targets SET domain methyltransferase activity to hox gene promoters. Mol Cell 2002;10:1107–17. Nakamura T, Mori T, Tada S, Krajewski W, Rozovskaia T, Wassell R, et al. ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol Cell 2002;10:1119–28. Hess JL. MLL: a histone methyltransferase disrupted in leukemia. Trends Mol Med 2004;10:500–7. Krivtsov AV, Armstrong SA. MLL translocations, histone modifications, and leukaemia stem-cell development. Nat Rev Cancer 2007;7: 823–33. Okada Y, Feng Q, Lin Y, Jiang Q, Li Y, Coffield VM, et al. hDOT1L links histone methylation to leukemogenesis. Cell 2005;121:167–78. Bitoun E, Oliver PL, Davies KE. The mixed-lineage leukemia fusion partner AF4 stimulates RNA polymerase II transcriptional elongation and mediates coordinated chromatin remodeling. Hum Mol Genet 2007;16:92–106. www.aacrjournals.org 24. Mueller D, Bach C, Zeisig D, Garcia-Cuellar MP, Monroe S, Sreekumar A, et al. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood 2007;110:4445–54. 25. Zhang W, Xia X, Reisenauer MR, Hemenway CS, Kone BC. Dot1a-AF9 complex mediates histone H3 Lys-79 hypermethylation and repression of ENaCalpha in an aldosterone-sensitive manner. J Biol Chem 2006;281:18059–68. 26. Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell 2011;20:66–78. 27. Milne TA, Martin ME, Brock HW, Slany RK, Hess JL. Leukemogenic MLL fusion proteins bind across a broad region of the Hox a9 locus, promoting transcription and multiple histone modifications. Cancer Res 2005;65:11367–74. 28. Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 2011;20:53–65. 29. Basavapathruni A, Jin L, Daigle SR, Majer CR, Therkelsen CA, Wigle TJ, et al. Conformational adaption drives potent, selective and durable inhibition of the human protein methyltransferase DOT1L. Chem Biol Drug Des 2012;80:971–80. 30. Yu W, Chory EJ, Wernimont AK, Tempel W, Scopton A, Federation A, et al. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat Commun 2012;3:1288. 31. Marango J, Shimoyama M, Nishio H, Meyer JA, Min DJ, Sirulnik A, et al. The MMSET protein is a histone methyltransferase with characteristics of a transcriptional corepressor. Blood 2008;111:3145–54. 32. Li B, Jackson J, Simon MD, Fleharty B, Gogol M, Seidel C, et al. Histone H3 lysine 36 dimethylation (H3K36me2) is sufficient to recruit the Rpd3s histone deacetylase complex and to repress spurious transcription. J Biol Chem 2009;284:7970–6. 33. Chesi M, Nardini E, Lim RS, Smith KD, Kuehl WM, Bergsagel PL. The t (4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood 1998;92:3025–34. 34. Keats JJ, Maxwell CA, Taylor BJ, Hendzel MJ, Chesi M, Bergsagel PL, et al. Overexpression of transcripts originating from the MMSET locus characterizes all t(4;14)(p16;q32)-positive multiple myeloma patients. Blood 2005;105:4060–9. 35. Keats JJ, Reiman T, Maxwell CA, Taylor BJ, Larratt LM, Mant MJ, et al. In multiple myeloma, t(4;14)(p16;q32) is an adverse prognostic factor irrespective of FGFR3 expression. Blood 2003;101:1520–9. 36. Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet 2010;42: 181–5. 37. Bodor C, Vera G, Kohlmann A, Tan K, Okosun J, Popov N, et al. High incidence of EZH2 mutations with variable mutation load in follicular lymphoma and its consequences for EZH2 targeted therapy[abstract]. In Proceedings of the 54th ASH Annual Meeting and Exposition; 2012 Dec 8–11; Atlanta, GA. Washington, DC: ASH; 2012. Abstract nr 545. 38. Sneeringer C, Scott MP, Kuntz K, Knutson SK, Pollock RM, Richon V, et al. Coordinated activities of wild-type plus mutant EZH2 drive tumorassociated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci U S A 2010;107: 20980–5. 39. Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol 2012;8:890–8. 40. McCabe MT, Graves AP, Ganji G, Diaz E, Halsey WS, Jiang Y, et al. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc Natl Acad Sci U S A 2012;109:2989–94. 41. Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer 2011;11:481–92. 42. Ginn KF, Gajjar A. Atypical teratoid rhabdoid tumor: current therapy and future directions. Front Oncol 2012;2:114. Clin Cancer Res; 19(23) December 1, 2013 Downloaded from clincancerres.aacrjournals.org on December 3, 2013. © 2013 American Association for Cancer Research. 6349 Published OnlineFirst August 19, 2013; DOI: 10.1158/1078-0432.CCR-13-0223 Copeland 43. Wilson BG, Wang X, Shen X, McKenna ES, Lemieux ME, Cho Y-J, et al. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 2010;18:316–28. 44. Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, et al. Durable tumor regression in genetically altered malignant rhab- 6350 Clin Cancer Res; 19(23) December 1, 2013 doid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A 2013;110:7922–7. 45. Daigle SR, Ohava EJ, Therkelsen CA, Basavapathruni A, Jin L, BoriackSjodin A, et al. A DOT1L inhibitor blocks MLL-fusion leukemia. Blood 2013;122:1017–25. Clinical Cancer Research Downloaded from clincancerres.aacrjournals.org on December 3, 2013. © 2013 American Association for Cancer Research.