Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
The RA signaling pathway in early
neural tube development
A potentially interesting pathway
for toxic risk assessment
and AOP construction
Rianne Jansen, 3180840
RIVM, Bilthoven
August, 2013
Master thesis for the master Cancer Genomics and Developmental Biology at the University of
Utrecht.
Supervisors:
Prof. Dr. Aldert Piersma, Dr. Ilse Tonk
UU examiner:
Prof. Dr. Blaauboer
1
Table of Contents
Page
Introduction
Neural tube formation and patterning
Patterning along the antero-posterior and dorso-ventral axis
Retinoic acid
Retinoic acid synthesis and degradation
3
4
6
8
9
Retinoic acid and neural tube development
10
Gradients of RA and FGF control neuronal differentiation, anterior-posterior patterning of
the neural tube and axis elongation
10
Collinear Hox gene expression provides the neural tube with positional identity along the
anterior-posterior axis
13
RA and Patterning of the hindbrain
16
Regulation of RA activity in the hindbrain
18
RA activates Hox gene expression in the hindbrain in a sequential manner
21
Initial anterior-posterior identity in the hindbrain might be provided by RA induced
expression of Hox genes
22
Examples of compounds affecting neural tube development
Ethanol
Ethanol competes for RALDH2 activity during gastrulation stages
Ethanol exposure changes RA levels in the hippocampus and cortex during late
embryonic development as well as in the adult brain
Ethanol exposure results in increased RA synthesis and altered RA signaling in the
developing cerebellum
The effect of Ethanol exposure on RA signaling is stage dependent
Triazoles
Triazoles interfere with RA signaling
Triazoles inhibit CYP26 activity
A potential mechanism for RA mediated teratogenic effects of triazoles
23
23
24
26
27
27
28
28
29
30
Conclusions
33
Acknowledgements
36
References
36
2
Introduction
During embryonic development tight regulation of developmental processes is very important. When
the finely balanced regulation is disturbed, for example by a genetic mutation or exposure to a toxic
compound, this may result in aberrant development. One very important process during development
is the formation and patterning of the neural tube, the precursor of the central nervous system. It is
therefore important to be able to assess whether a compound, for example a drug or a compound
present in the environment, has the potential to disturb the fine balance and thus may affect neural
tube development.
The classic way to test for possible teratogenic effects of compounds is by exposing model
organisms (animals) to a high dose of the compound to see if development is affected (Krewski et
al., 2007). However this requires a high amount of test animals, which is ethically undesirable, it is
time consuming and expensive, and it gives only limited insight in the stage and tissue dependent
mechanisms underlying the toxic effects. Therefore there is a demand for alternative approaches.
In modern toxicity research, novel approaches are being developed for assessment of potential risks.
One of these approaches is based on the idea of studying the effect of compounds on specific key
pathways, of which its distortion may be indicative of an adverse effect on (for example) embryonic
development. Determining whether a compound has an effect on these pathways may therefore
provide information on potential teratogenic effects. When such tests are done in cell lines, for
example in ES cells differentiated into a certain cell lineage (e.g. neural embryonic stem cells), it
provides the opportunity to do an initial assessment of potential toxic risks on a large scale for many
compounds at once in a relatively short time (Theunissen et al., 2013). If it is known which key
pathways are altered and how they are altered, this will give an indication of which developmental
processes might be affected and what the effect may be. It thus becomes possible to predict what the
potential risks are of exposure to the compound. In other words, it becomes possible to construct a
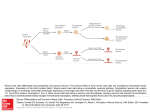
putative adverse outcome pathway (AOP) for this compound. An AOP describes the series of events
linking the molecular initiating event (interaction of the compound with molecules within the cell) to
the adverse outcome for an individual or population (for example neural tube defects, embryonic
lethality) (fig. 1) (Ankley et al., 2010). Such an adverse outcome pathway may be dependent on the
life stage during which the individuals are exposed; a compound might have quite a different effect
on the developing embryo than on an adult individual.
Fig. 1 An adverse outcome pathway describes the sequence of events that links a key molecular initiating event
to an adverse outcome on the organism or population level. A toxic compound interacts with a biological target. In
turn, this may lead to an effect on cellular level and subsequently on the organ level, which may produce an adverse
outcome on the organism and population level. After: Ankley et al., 2010.
3
In the case that a key pathway is used as a molecular read-out for a toxic effect, for example the RA
pathway as a read-out for potential toxic effects on neural tube development, the AOP can be
partially constructed, linking the toxic compound to the cellular responses (alterations of this key
pathway and its targets) which could then be linked to effects on the organ level (altered neural tube
development) and to organismal responses (lethality, impaired development, etc.).
A challenge is identifying such key pathways. These pathways have to be (at least) predictive of
toxic effects. A potentially interesting pathway in relation to neural tube development is the retinoid
signaling pathway, as RA signaling plays an important role in patterning of the embryo and
differentiation. It is involved in several processes relating to neural tube development, for example
anterior-posterior patterning of the neural tissue, patterning of the hindbrain and dorso-ventral
patterning of the spinal cord (reviewed by Maden, 2005 and Rhinn and Dollé, 2012). Furthermore
aberrant RA signaling, either excess or absence thereof, is associated with neural tube defects
(Maden, 2006). Exposure to excess RA through maternal supplementation leads to neural tube
defects such as exencephaly and spina bifida, the exact effect on neural tube development is
dependent on time of exposure. Also mutation of RA catabolizing CYP26 enzymes causes similar
neural tube defects (among other defects). Absence of RA signaling, either through mutation of RA
producing enzymes or its receptors or by a vitamin A deficiency, results in neural tube defects as
well (exencephaly and/or spina bifida). Because of the role of RA signaling in neural tube
development and its association with neural tube defects, studying whether a compound has an effect
on this pathway may give information about neurotoxicity of the compound.
In this paper, I will review the role of retinoid signaling during neural tube development, the
pathway(s) through which RA exerts its effect, with its time and space specific context, and I will
propose a set of genes associated with this pathway that may be used to characterize (potential)
teratogenic effects of compounds on neural tube development. Additionally, I will give two
examples of compounds that are known to interfere with RA signaling and cause neural tube defects
and I will propose an AOP for each of these compounds.
Neural tube formation and patterning
The formation and patterning of the neural tube involves many different steps and processes. Here I
will give a short overview of these steps, as described by Wolpert et al (2007) and Dias and
Partington (2004). The very first step towards development of the nervous system is the induction of
neural ectoderm from the dorsal ectoderm in response to signals from the node and notochord
(“neural induction”) (Wolpert et al., 2007; Dias and Partington, 2004). The dorsal ectoderm thickens
to form the neural plate and thereby becomes morphologically different from the surrounding
epithelium. In humans, this thickened neural ectoderm becomes visible around postovulatory day 16
(Dias and Partington, 2004). The neural plate becomes longer and narrower, by a process called
convergent extension (Wallingford et al., 2013). At the same time the lateral edges of the neural plate
start to fold upwards (dorsally) and eventually the edges meet at the dorsal midline and fuse to form
the hollow neural tube (fig. 2) (Wolpert et al., 2007; Dias and Partington, 2004). The neural
ectoderm then separates from the rest of the ectoderm, the future epidermis, which now overlies the
4
neural tube. The neural crest cells dissociate from the edges of the neural plate and migrate to their
destination. In humans formation of the neural folds occurs between postovulatory day 19 and 21
(Dias and Partington, 2004), in mice around E8 (Wolpert et al., 2007). Closure of the human neural
tube occurs between the third and fourth week of gestation, in mice between E8.5 and E10
(Wallingford et al., 2013; Ybot-Gonzalez et al., 2002).
Fig. 2 Neural tube closure in the vertebrate embryo.
A: In the ectoderm of the embryo the dorsal ectoderm thickens to form the neural plate. The lateral edges of the neural
plate start to fold upwards and then towards the midline. When the edges meet, they fuse to form a hollow tube. The
neural crest cells dissociate and migrate to their destination.
B: Closure does not occur along the whole length of the neural plate at once, but starts around the caudal
hindbrain/rostral cervical levels and from there the closing movement extends further in anterior and posterior direction.
At the same time the neural tube elongates at the posterior end as the node regresses. The anterior and posterior
neuropores have not yet closed in the fourth picture of the embryo. Figure from: Wolpert et al., 2007 (fig. 7.34);
Wallingford et al., 2013.
Folding of the neural tube does not occur along the whole length of the neural plate at once, but it
occurs in waves (Dias and Partington, 2004). In human embryos the first wave of closing starts in the
region of the caudal hindbrain or rostral spinal cord, and then continues to extend in anterior and
posterior direction (fig. 3). The ends of the neural tube still remain open and form the anterior and
posterior neuropore. Later, the tube closes at the anterior and posterior end as well (Dias and
Partington, 2004; Wolpert et al., 2007). Failure to properly close the neural tube can lead to various
defects such as exencephaly (failure to close cranially) anencephaly (failure to close cranially) and
meningomyelocele (failure to close caudally), better known as spina bifida (Dias and Partington,
2004; Wallingford et al., 2013; Maden, 2006). Failure to close along the whole A-P axis is referred
5
to as craniorachischisis, one cause of which can be disruption of the lengthening and narrowing of
the neural plate by convergent extension (Wallingford et al., 2013). All conditions are lethal, except
for meningomyelocele (Wallingford et al., 2013). That process of neural tube closure is a sensitive
process, is demonstrated by the fact that it is the second most common birth defect (Wallingford et
al., 2013).
Fig. 3 Neural tube closure occurs in waves. The first wave starts in the region of the caudal hindbrain or rostral
spinal cord and then extends in an anterior and posterior direction. Failure to close at the anterior end (wave 2) results in
anencephaly, while failure to close at the posterior end results in meningomyelocele (spina bifida). The last portion of the
spinal cord is formed by a different process called secondary neurulation. This part develops as a solid rod, which then
develops a cavity. Figure from: Gilbert, 2000
The caudal-most part of the neural tube results from a different process, called secondary
neurulation. This part is produced by the caudal cell mass (also known as posterior growth zone in
the tail bud of the embryo), an area containing pluripotent cells. The caudal cell mass produces an
initially solid rod, which later develops a cavity (Dias and Partington, 2004). This part fuses with the
rest of the neural tube that derived from the neural plate. In humans the portion produced by
secondary neurulation is only small (Dias and Partington, 2004).
Patterning along the antero-posterior and dorso-ventral axis
At many stages of neural tube development signals from other tissues are important, such as signals
from the node, notochord, overlying ectoderm, and somites (e.g. RA).
The very first step towards development of the nervous system is the induction of neural ectoderm
from the dorsal ectoderm in response to signals from the node and notochord. Initially BMP proteins
are expressed throughout the ectoderm; inhibition of BMPs in the dorsal ectoderm allows this tissue
to become neural ectoderm. BMP antagonists such as noggin, follistatin and chordin are important
for the induction of neural tissue, as well as FGF (Wolpert et al., 2007; Dias and Partington, 2004).
Signals from the mesoderm help pattern the neural tissue along the anterior-posterior (A-P) axis,
after its induction. It is thought that all neural tissue is first specified as anterior neurectoderm after
neural induction. Posteriorizing signals will then change the identity of the neural tissue (Wolpert et
al., 2007; Dias and Partington, 2004). WNTs, RA and FGFs are important signals in this respect as
they impose posterior identity in a dose dependent manner, inducing the most posterior identity in
the caudal tissue (Wolpert et al., 2007; Dias and Partington, 2004). This A-P patterning is reflected
by region specific expression patterns of the Hox genes and other homeobox genes (Dias and
6
Partington, 2004), which is important for proper further region specific development of the neural
tube. Along the A-P axis the neural tube develops into several distinct regions: the forebrain,
midbrain, hindbrain and spinal cord, which will develop and regionalize further. The hindbrain for
example will segment, forming eight rhombomeres (fig. 4). Each rhombomere will give rise to
specific cranial ganglia and/or motorneurons (Kiecker and Lumsden, 2005).
Fig. 4 The hindbrain is segmented into eight rhombomeres.
Each individual segment produces neurons innervating distinct areas of the embryo (Kiecker and Lumsden, 2005). On
the left the positions of the sensory cranial ganglia is depicted. On the right three sets of motor neurons are depicted that
innervate the branchial arches, which will innervate the head and face. A fourth set contributes to the vagus nerve. ov =
otic vesicle, green arrow indicate migration of neural crest cells into the branchial arches. Figure from: Kiecker and
Lumsden, 2005.
The neural tube is also patterned along the dorso-ventral axis by two opposing signals (fig. 5). Sonic
hedgehog (SHH) produced by the notochord, and later also by the floor plate, specifies the neural
ectoderm as ventral (Wolpert et al., 2007; Dias and Partington, 2004). SHH also induces the
formation of the floor plate itself (Dias and Partington, 2004). BMPs produced by the non-neural
dorsal (epidermal) ectoderm, which overlies the neural tube after fusion of the neural folds, specifies
the dorsal side (Wolpert et al., 2007; Dias and Partington, 2004). Later, the roof plate produces
BMPs as well. Different types of neurons will differentiate in different regions along this D-V axis
(Wolpert et al., 2007; Dias and Partington, 2004).
7
Fig. 5 Opposing signals from the notochord and surface ectoderm pattern the neural tube along the dorsalventral axis. The notochord secretes SHH and the surface ectoderm BMPs. Later the floor plate starts to express SHH as
well, and the roof plate expresses BMP4 and other TGFβ family members. SHH diffuses from the ventral midline and
patterns the ventral spinal cord, BMPs diffuses from the dorsal midline. SHH and BMPs antagonize each other’s effects.
Figure obtained from: Gilbert (2010)
Retinoic acid
RA is a small, vitamin A derived molecule. RA has many roles in many processes during embryonic
development. The response to RA is dependent on the context, both in time and space: the effect can
differ between types of tissues and between different stages during development. In general, RA
signaling is involved in patterning of the embryo, and with respect to anterior posterior patterning it
is a posteriorizing factor. Furthermore it induces differentiation (as opposed to proliferation). During
embryonic development RA is involved in many processes, among which are: A-P patterning of the
embryo, protecting the somites from left-right specific signaling, thus maintaining bilateral
symmetry, and limb development, but it is also involved in several stages of neural tube development
(reviewed by Rhinn and Dollé, 2012). It is therefore a very important signaling molecule.
RA is synthesized from gastrulation stages onward in the primitive streak and mesodermal cells and
later in presomitic and somitic mesoderm (Rhinn and Dollé, 2012). After synthesis it can then diffuse
across tissues, forming a gradient. The main source of retinoids for the embryo is maternal retinol,
transferred across the placenta, however RA seems able to cross the placenta as well as RA
supplementation of the mother does affect the embryo (Rhinn and Dollé, 2012). In the target cells,
RA diffuses across the cell membrane and then binds to a nuclear receptor, transforming the receptor
from a transcriptional repressor into an activator. It thereby activates the expression of target genes.
RA can directly regulate its own activity through feedback loops. For example, it can regulate
expression of its own receptors and of Cyp26a1, which encodes an RA catabolizing enzyme
(reviewed by Rhinn et al., 2012). RA seems to diffuse into the neural tube with a higher preference
than into other tissues (Maden, 2006).
There are several RA receptors RARα, RARβ and RARγ which can form heterodimers with retinoid
X receptors RXRα, RXRβ and RXRγ (reviewed by Rhinn and Dollé, 2012). RARα, RXRα and
RXRβ are broadly expressed in many tissues, whereas the other receptors have more tissue specific
expression patterns. The receptors bind to RA recognition elements (RAREs) in the promoters of
target genes. If no RA is bound, these receptors repress gene expression, but after binding they turn
into an activator (Rhinn and Dollé, 2012). The receptors form dimers, and binding of RA to one of
8
the two receptors is sufficient to activate them. RA might also bind to other nuclear receptors such as
PPARβ or PPARγ (Rhinn and Dollé, 2012). Besides their role in regulation of expression, RA
receptors might also have cytoplasmic roles (Kumar et al., 2010).
Retinoic acid synthesis and degradation
The synthesis and degradation pathways of RA have been well described. Below is a short overview,
based on the review by Rhinn and Dollé (2012), unless otherwise indicated.
The source of RA is vitamin A (retinol), which must be obtained through diet. Several enzymes are
involved in the localized synthesis of RA from retinol (fig. 6A). The first step is the conversion of
retinol into retinaldehyde. This reaction is catalyzed by alcohol dehydrogenases (ADHs) or retinol
dehydrogenases (RDHs), the main enzyme catalyzing this reaction during development being
RDH10 (Rhinn and Dollé, 2012). In some tissues STRA6 can facilitate retinol uptake.
In a second step retinaldehyde is oxidized (oxidation) to retinoic acid by retinaldehyde
dehydrogenases (RALDH1, RALDH2, RALDH3). This is mainly done by RALDH2, as RALDH2 is
the earliest and most widely expressed molecule. RALDH2 is first expressed in the primitive streak,
node and mesodermal cells. Later, it is expressed in the presomitic and somitic mesoderm and
anterior forebrain (Rhinn and Dollé, 2012). In mice, RALDH1 and 3 are expressed only after day
E8.5 in the eyes and olfactory system. After synthesis, retinoic acid can diffuse to other tissues,
forming a gradient. Intracellularly RA can be bound to cellular retinoic acid-binding proteins
CRABP1 and CRABP2.
Fig. 6 RA synthesis and degradation.
A: Retinol dehydrogenases and aldehyde dehydrogenases, most importantly RDH10, catalyze the reaction of retinol into
retinaldehyde. Retinaldehyde is further oxidized to retinoic acid by retinaldehyde dehydrogenases (RALDH), mainly by
RALDH2. B: RA is degraded by the action of CYP26 enzymes.
Figure is adapted from: Rhinn and Dollé, 2012.
RA can be degraded by the CYP26 enzymes (CYP26A1, CYP26B1 and CYP26C1) (Rhinn and
Dollé, 2012). They convert RA into 4-hydroxy-RA and 4-oxo-RA (fig. 6B). Tissue specific
expression of CYP26 enzymes protects these tissues from the influence of RA. Cyp26a1 is for
example expressed in the tail bud which contains the stem cells. Keeping this region free of RA
prevents induction of differentiation by RA and ensures that this stem cell region is maintained
(Wilson et al., 2009). RA is able to induce expression of its own catabolizing enzymes.
9
Retinoic acid and neural tube development
Retinoic acid has multiple functions during neural tube development (reviewed by Rhinn and Dollé,
2012). For instance, it plays an important role in anterior-posterior patterning of the neural plate and
tube, specifically of the spinal cord and caudal hindbrain, and in neural differentiation. This anteriorposterior patterning of the embryo and the onset of differentiation are tightly coupled to the process
of axis elongation (fig. 7). Furthermore, RA is needed for dorsal-ventral patterning of the neural
tube. Studies suggest a role for RA in patterning of the forebrain as well. At later stages of brain
development RA is expressed in the developing hippocampus, cortex and cerebellum. A function of
RA in the hippocampus seems to persist into adulthood. In the adult brain, it may also have a
function in the forebrain/cortex.
Gradients of RA and FGF control neuronal differentiation, anterior-posterior
patterning of the neural tube and axis elongation
Patterning of the embryo is coupled to the morphogenetic movements that elongate the embryo (fig.
7). As the node regresses, tissue is left behind and thereby the embryonic axis gets elongated at its
caudal end. As the node moves away (posteriorly) the cells in this tissue differentiate. The interplay
between factors produced in the caudal-most region in and around the node, such as FGF8 and WNT
proteins, on the one hand and RA produced by the somites on the other hand are important for the
regulation of the onset of differentiation and for the patterning along the A-P axis of both the
mesoderm (e.g. somites) and neural ectoderm (Wilson et al., 2009). FGF8 maintains the proliferative
capacity of the cells in the caudal region (which allows elongation of the embryo) and suppresses
differentiation, while RA promotes differentiation (Rhinn and Dollé, 2012). Fgf8 mRNA is produced
in and around the node. As Fgf8 is gradually degraded in the cells leaving the node region (anterior
to the node), an Fgf8 mRNA gradient arises with a high concentration of FGF8 in the node that
decreases in anterior direction, resulting in a gradient of FGF8 protein (Wolpert et al., 2007). RA is
formed in the somites and anterior presomitic mesoderm and can diffuse from there to neighboring
tissues, such as the neural ectoderm. As the node moves is further posteriorly, the tissue that is left
behind is subjected to gradually lower FGF8 levels and higher RA levels. This drop in FGF8 and the
presence of RA are necessary to promote differentiation of the cells in the mesoderm and neural
ectoderm. RA seems to favor neural differentiation above mesodermal differentiation, as excess RA
leads to formation of neural tissue at the expense of paraxial mesoderm (Wilson et al., 2009). It also
leads to axis truncation, probably due its differentiating effect on the stem cells in the node.
Furthermore RA induces differentiation into the neural lineage in mouse embryonic stem cells
(Rhinn and Dollé, 2012). Coupled to the regulation of neural differentiation, is also regulation of
dorso-ventral patterning genes. FGF8 suppresses Shh expression in the floor plate, while RA
promotes its expression, thereby controlling onset of ventral patterning (Diez del Corral et al., 2003;
Wilson et al., 2009). At the same time FGF, WNTs and RA are also important for patterning of the
tissue along the A-P axis. This patterning is reflected by specific expression patterns of Hox genes in
10
neural and mesodermal tissue (Deschamps and Van Nes, 2005). RA is especially important for
patterning of the hindbrain and anterior spinal cord.
Fig. 7 Gradients of RA and FGF control axis elongation, (neuronal) differentiation, anterior-posterior
patterning and dorso-ventral patterning of the neural tube.
A: Fgf8 is produced in the node. As cells leave the node area during axis elongation, the Fgf8 levels in these cells
decrease. As a result, cells further away from the node will experience lower FGF8 levels. FGF8 thus forms a gradient in
the posterior embryo. RA produced by the somites diffuses in anterior and posterior direction, forming a gradient as well.
B: RA and FGF8 signaling repress each other. Together RA and FGF8 regulate the process of axis elongation, the onset
of neural differentiation, anterior-posterior (A-P) patterning and dorso-ventral (D-V) patterning of the embryo. Adapted
from: Deschamps and Van Nes, 2005.
Fgf8 and RA negatively regulate each other’s activity (fig. 8). Thereby the proliferative area in and
around the node is kept free of RA, while the presence of RA and the drop of Fgf8 levels allow for
differentiation anterior to the node. In mice the expression of Fgf8 is induced by WNT3a (reviewed
by Wilson et al., 2009). FGF8 in turn suppresses RA signaling, to prevent RA from inducing
differentiation; in chick FGF8 has been shown to suppress Raldh2 expression in the presomitic
mesoderm and also the expression of Rarb in the neural ectoderm (although this could be an indirect
effect of lower RA signaling: Rarb has a RARE and can thus be regulated by RA), in mice it
stimulates expression of Cyp26a1 in the caudal region, which effectively clears this region of RA
(reviewed by Wilson et al., 2009). In turn, RA produced by the somites inhibits Fgf8 and Wnt3a
expression (Wilson et al., 2009).
11
Fig. 8 FGF and RA regulate each other’s activity.
A: molecular interactions observed in chick. B: molecular interactions observed in the mouse.
FGF8 inhibits RA synthesis and signaling. RA in turn represses Fgf8, but stimulates CYP26a expression in the caudal
embryo. Wnt8c is stimulated by Fgf8 signaling. WNT8C mediates the transition from FGF8 to RA signaling. When Fgf8
levels are sufficiently low, WNT8C can induce Raldh2 expression. Eventually Wnt8c expression is lost due to loss of
stimulation by Fgf8 and repression by RA. Fgf8 inhibits Shh expression in the floor plate, while RA promotes it. Figure
from: Wilson et al., 2009
The transition from FGF to RA signaling is probably mediated by WNT8 proteins (Olivera-Martinez
and Storey, 2007). In chick, FGF signaling stimulates Wnt8c expression (ortholog of Wnt8a in the
mouse). WNT8c in turn is able to promote expression of Raldh2 in vitro. In vivo, lifting the
inhibition by FGF of Raldh2 by itself does not elevate RALDH2 levels, nor does stimulation of
canonical Wnt signaling, but when FGF is blocked and Wnt signaling is increased Raldh2 expression
is elevated. Similarly when Wnt signaling is inhibited, the onset of Raldh2 expression is inhibited.
This shows that both derepression of Raldh2 by FGF and stimulation of Raldh2 by Wnt are needed
to induce Raldh2 expression. As WNT8c is the only caudal Wnt signal promoted by FGF and only
WNT8c is expressed near the domain of Raldh2 expression onset, it is probable that it is indeed
WNT8c that mediates these Wnt specific effects. Thus WNT8c stimulates Raldh2 expression, but
only if FGF levels have sufficiently decreased (Olivera-Martinez and Storey, 2007). WNT8c
expression declines less rapidly than FGF8, so there is a region where Wnt signals are present
without FGF8. Here Raldh2 expression can be stimulated. As FGF8 is needed to maintain WNT8c
expression, eventually WNT8c expression is lost as well when FGF8 signaling is attenuated. Both
FGF and WNT8c are able to repress neural differentiation through different mechanisms, as
demonstrated by their inhibitory effects on Neurogenin1 and NeuroM expression, however Wnt
signaling is less efficient. In the developing embryo the decrease of FGF and Wnt signaling and the
presence of RA allow for differentiation.
12
Collinear Hox gene expression provides the neural tube with positional identity
along the anterior-posterior axis
As discussed in the previous section, RA is needed for neural differentiation throughout the
developing spinal cord and this process of differentiation is tightly coupled to the process of axis
elongation, by which the embryonic tissues –including the spinal cord- are extended posteriorly. Wnt
signals, FGFs and RA together impose anterior-posterior patterning of the neural plate. FGF and RA
modulate the expression patterns of the Hox genes in the neural ectoderm of the future spinal cord
and posterior hindbrain (also in the mesoderm, although the anterior expression boundaries are
different between the mesoderm and neural ectoderm) (Young and Deschamps, 2009). The Hox
genes are activated in sequential order: the first Hox genes within a Hox cluster (paralogous group 1
at the 3’end) are expressed first and most anteriorly, while the more 5’ genes are activated later in
progressively more posterior tissues (fig. 9) as these are formed cells leaving the regressing node.
Thus, Hox genes are expressed in a sequential order (1-13) along the A-P axis and in time, which is
referred to as spatio-temporal collinearity. Anteriorly their expression domains have a sharp
boundary, but in the posterior direction their expression gradually decreases. The expression domains
of the neighboring Hox genes in a cluster do overlap and the combination of Hox genes expressed at
a certain level along the A-P axis determines the axial identity of the tissue (fig. 9). The tissue will
then further develop into structures corresponding to the acquired identity. Signals from the node,
mainly of FGFs, largely determine this specific Hox gene expression. Nodes from older embryos,
thus from a more caudal position, induce Hox gene expression typical of increasingly posterior
tissues (Liu et al. ,2001).
These specific Hox gene expression patterns provide the neuronal cells with a positional identity
along the A-P axis. A correct Hox gene expression pattern is essential for neural development and
positional identity of the neurons, as demonstrated by defects exhibited by Hox mutant mice
(reviewed by Young and Deschamps, 2009) and the effects of modulating Hoxc6 and Hoxc9
expression on motor neuron identity (Dasen et al., 2003). For instance, correct expression is
important for projection of both the afferents of sensory neurons and of the axons of motor neurons
(Liu et al., 2001; Young and Deschamps, 2009).
13
Fig. 9 Collinear Hox gene expression in the neural tube and mesoderm provides the tissue with identity along
the anterior-posterior axis.
The genes within a Hox cluster are expressed in a spatially and temporally collinear manner: the 3’ genes are expressed
first and in the most anterior structures, 5’genes are expressed progressively later in progressively more posterior tissue.
A: Hox expression in the neural tube and mesoderm the mouse embryo. The anterior expression boundaries of the Hox
genes differ between the neural tube and mesoderm. The expression of the Hox genes extends more anteriorly in the
neural ectoderm, and this is dependent on RA signaling. The combination of Hox genes expressed at a certain level along
the A-P axis determines the axial identity of the tissue and this results further development of this tissue according to its
acquired identity.
B: Collinear Hox expression of Hoxc5-Hoxc10 in the spinal cord of HH stage 24 embryos. Fig. 9A downloaded from:
http://www.pbs.org/wgbh/nova/genes/fate-04.html; fig. 9B: Liu et al., 2001.
It is thought that FGFs, notably FGF8, produced by the node induce posterior Hox gene expression
in the spinal cord in a concentration dependent manner (fig. 10) (Liu et al., 2001; Bel-Vialar et al.,
2002; Martinez and Storey, 2007). This is likely in part mediated through the family of CDX proteins
(Bel-Vialar et al., 2002). When the cells leave the node region, they have already acquired a caudal
Hox code under influence of FGF (Liu et al., 2001). Experiments in chick suggest that RA signaling
refines this expression pattern by inducing expression of the more anterior Hox genes in the rostral
spinal cord (Liu et al., 2001; Bel-Vialar et al., 2002; Martinez and Storey, 2007). However, in E7.75
Raldh2-/- mouse embryos early Hoxb1 expression is unaffected (Niederreither et al. 2000); only later
its expression is disturbed, suggesting that the early expression of this 3’ Hox gene is not dependent
on RA. Similarly, Hox4 genes are expressed in the neural tissue of the Raldh2-/- embryo, although
the anterior expression boundary differs from wildtype in E8.25 embryos (Niederreither et al. 2000).
Thus only at a later stage, RA functions to extend the anterior boundary of the expression domains of
the anterior and central Hox genes (Hoxb1-Hoxb8) in the neural ectoderm to their definitive, more
anterior location in the posterior hindbrain rather than that it is responsible for the onset of these Hox
genes (fig. 10) (Oosterveen et al., 2003; reviewed by Young and Deschamps, 2009).
The Hox clusters contain RAREs, so they can be directly regulated by RA (Oosterveen et al., 2003).
At first, the induced caudal Hox genes are not sensitive to RA yet, but later they do become sensitive
in a sequential order: the first to be expressed (3’) Hox genes become sensitive to RA first (before
E8.5), then the later induced, more 5’ Hox genes become sensitive (e.g. Hoxb8 at E10.5) (Gould et
al., 1998; Oosterveen et al.; reviewed by Deschamps and Van Nes, 2005).
14
Fig. 10 FGF8 and RA regulate Hox gene expression in the neural tube
FGF8 establishes collinear Hox gene expression patterns in the neural tube. Supposedly, increasing FGF8 levels in the
node induce increasingly posterior Hox gene expression during axis elongation. At later stages GDF11 enhances the
posteriorizing effects of FGF8. Later, RA modulates Hox gene expression patterns. In response to RA the expression
domain of the Hox genes expands anteriorly, shifting the anterior limit of expression anteriorly. The 3’ Hox genes
become sensitive to RA first, later the more 5’ Hox genes become responsive. The specific Hox gene expression pattern
provides the cells of the neural tube with positional identity along the rostro-caudal axis. This is necessary for correct
projection of efferents and afferents of neurons in the spinal cord, and for correct rhombomere specification in the
hindbrain.
One way the node could be able to induce the expression of different Hox genes during axis
elongation, is by increasing FGF levels while it moves posteriorly. Indeed, increasing levels of FGF
are able to stimulate increasingly posterior Hox expression (Liu et al., 2001; Bel-Vialar et al., 2002)
and in situ hybridization experiments do suggest FGF levels in the node rise over time (Liu et al.,
2001). Since in situ hybridization experiments are not quantitative the latter should be confirmed.
Another possibility is that the Hox gene expression is dependent on the time spent in the node, and
thus the duration of exposure to FGF signaling1*. Furthermore, from the 11 somite stage Gdf11 on
becomes expressed in the tail bud of the chick embryo and in vitro GDF11 is able to enhance the
1
Both alternatives are possible: older nodes are able to induce more posterior hox gene expression in neural tissue (Liu et
al., 2001). This suggests that it is the strength of the signals from the node that give the neural tissue its identity, as these
neural cells themselves did not spend a longer time in the node and thus weren’t exposed to FGF for a longer time. At
first glance this might point to a model in which rising FGF levels in the node cause increasingly more posterior (5’) Hox
gene expression. On the other hand, it could be that the neural progenitor cells in the node do acquire an older, more
caudal profile, with concomitant Hox gene expression, and signal to the neighboring grafted tissue imposing the older
identity on the tissue.
15
posteriorizing effects of FGF, but it is not able to affect Hox gene expression on its own. Thus, in the
caudal spinal cord the posteriorizing action of FGFs is aided by GDF11 to cause even more posterior
Hox gene expression.
RA and Patterning of the hindbrain
After closure of the neural tube in the hindbrain region, the hindbrain segments to form seven
(zebrafish) to eight rhombomeres (mouse, chick, human) (Rhinn and Dollé, 2012). Retinoic acid
produced by the somitic mesoderm plays an important role in the patterning of the posterior
hindbrain, where it is needed for correct, rhombomere specific, expression of Hox genes and other
transcription factors (Niederreither et al., 2000; reviewed by Deschamps and Van Nes, 2005). More
specifically, it is important for posteriorization of the caudal hindbrain rhombomeres, as
demonstrated by the effects of either absence of RA signaling or excess RA (fig. 11) (as reviewed by
Rhinn and Dollé, 2012). In absence of retinoic acid signaling, for example by vitamin A deficiency
or knocking out Raldh2, the tissue of the posterior hindbrain adopts a more anterior fate, the caudal
hindbrain is decreased in size and segmentation is impaired (for example Raldh2-/- phenotype: fig.
12) (Niederreither et al., 2000; Rhinn and Dollé, 2012). The vitamin A deficiency resulted in a loss
of cranial nerves IX, X, XI, and XII and associated sensory ganglia, and misguidance of nerves at
E12.5 (White et al., 1998; White et al., 2000). Similarly, interference with the RA receptors (by
compound knock-out thereof or by using a pan-RAR antagonist), leads to anteriorization of the
hindbrain as well (Rhinn and Dollé., 2012). Excess RA at the late gastrula or early neurula stages
leads to an anterior expansion of the hindbrain region at the expense of the anterior brain structures,
reflecting a posteriorization of these tissues. If excess RA is supplied at later stages, the
rhombomeres within the hindbrain undergo a posterior transformation (adopt a more posterior
identity). Experiments in which RA is blocked at progressively later stages suggest that RA patterns
the rhombomeres in a rostral to caudal order; for example blocking RA at an early stage results in
misspecification of r4-r8, while blocking it at later stages only results in defects at a progressively
more posterior level in the hindbrain (reviewed by Gavalas, 2002). Similarly, it seems that
increasingly higher RA levels are required for patterning of more posterior rhombomeres (reviewed
by Deschamps and Van Nes, 2005).
RA does not seem required for early patterning of the forebrain and formation of the mid-hindbrain
boundary (Niederreither et al., 2000), as this area is correctly specified in Raldh2-/- embryos,
although exposure to RA can impair development of these structures as excess RA leads to a
reduction of mid- and forebrain size due to expansion of the hindbrain area (Rhinn and Dollé, 2012).
In other words, part of the midbrain area has adopted a more posterior fate (hindbrain). Similarly,
loss of both Cyp26b1 and Cyp26c1 expression in the anterior hindbrain also leads a rostral shift of
the boundary between the mid- and hindbrain (Uehara et al., 2007).
16
Fig. 11 Altered RA signaling in the hindbrain results in hindbrain defects. RA is required for posteriorization of the
posterior hindbrain rhombomeres. Decreased RA signaling due to a vitamin deficient diet (VAD), knock-out of Raldh2,
or knock-out of the RA receptor leads to anteriorization of the hindbrain and a decrease of posterior hindbrain size. In
contrast, ectopic RA signaling, due to knock-out of Cyp26 genes leads to a posteriorization of the hindbrain
rhombomeres. Figure from: Rhinn and Dollé 2012.
Fig. 12 Due to absence of retinoic acid signaling in Raldh2-/- mutant mice the tissue of the posterior hindbrain
adopts a more anterior fate, the caudal hindbrain is decreased in size and segmentation is impaired.
RA seems required for posteriorization of the caudal hindbrain. In absence of RA, due to a knock-out of Raldh2,
specification of the anterior hindbrain rhombomeres (r1,r2) occurs relatively normally, whereas the caudal hindbrain is
anteriorized; it is expressing genes typical of more anterior rhombomeres and the expression of the genes typical of the
r5-r8 is decreased or lost (Niederreither et al., 2000). The caudal hindbrain is decreased in size and segmentation is
17
impaired. Gene expression patterns are depicted as observed at E8.25/E.8.5, before segmentation. The hindbrain defects
are depicted as observed at E9.5. Dark colors: high expression, lighter colors: lower expression. The pointy ends mean
that the expression boundary is not sharp. Figure from: Niederreither et al., 2000.
Regulation of RA activity in the hindbrain
Using mice with the RARE-LacZ reporter, it was shown that RA activity is transiently present up to
r2/r3 boundary; later the activity front regresses to the level of r4/r5 boundary (Sirbu et al., 2005). A
similar trend was observed in zebrafish (using RARE-fluorescent protein reporter): first RA activity
extends into the hindbrain and later it retreats again from the hindbrain (reviewed by Glover et al.,
2006). This RA pattern is established by diffusion of RA from the somites into the hindbrain and RA
degradation by dynamically expressed Cyp26 family members in the anterior part of the hindbrain.
There are however indications that there is another source of RA intrinsic in the hindbrain itself that
may contribute to this RA gradient (Niederreither et al., 2002; Mic et al., 2002).
Expression of CYP26 family members protects the anterior hindbrain against the posteriorizing
effects of RA (Rhinn and Dollé, 2012). Their expression changes over time and this dynamic
expression of CYP26 enzymes tightly controls RA activity within the rhombomeres in mouse, chick
and zebrafish. Although the exact expression profiles of the three different CYP26 enzymes differ
between the species, the basic principle is the same. The Cyp26 genes become expressed first in the
anterior hindbrain and later Cyp26 members become expressed in more posterior rhombomeres, with
each Cyp26 family member having its own rhombomere specific expression pattern. As a result of
this dynamic expression of these RA catabolizing enzymes, RA is excluded from progressively more
posterior rhombomeres, shifting the anterior boundary of RA activity posteriorly.
In chick, Cyp26a1 is initially expressed in the fore- and midbrain region. The expression domain
then regresses at the anterior end and at the posterior end it spreads out into hindbrain up to the r3/r4
border (of chick, after stage 8) (Blentic et al., 2003). Eventually its expression domain has shifted
such that it is only expressed in r3 (stages 9 and 10), after which its expression disappears. Cyp26c1
starts to be expressed in prerombomeres 1 and 2 just before stage 10, when formation of the
rhombomeres commences, then expands further posteriorly into the hindbrain. By stage 11 it is
expressed in r2, r3 and r5 and eventually it becomes restricted to r5 and r6 (Reijntjes et al., 2004).
Cyp26b1 becomes expressed in the (prospective) r4 and r6 from stage 7 onward (Reijntjes et al.,
2004).
A similar trend is observed for the mouse Cyp26 genes. Cyp26a1 is also first expressed in anterior
neural plate and from E7.5 on its expression domain expands as well (Sirbu et al., 2005). Eventually
its expression domain gets restricted to r2 at E8.5 (compared to r3 in chick) (Sirbu et al., 2005).
Cyp26c1 becomes expressed in r2 and r4 around E8 (Sirbu et al., 2005), after induction of Hoxb1 in
r4, and around or shortly after E8.5 expression in r4 is lost (Tahayato et al., 2003; Sirbu et al., 2005).
Cyp26b1 expression is first observed at E8 in prospective r3 and r5, with strongest expression in r5
(Maclean et al., 2001). By E9.5 Cyp26b1 expression has expanded, with its strongest expression
throughout r5 and r6 and additional expression in the ventral portion of r2-r4. This dynamic Cyp26
expression controls RA activity and causes RA to be cleared from progressively more posterior
hindbrain tissue, shifting the anterior boundary of its activity posteriorly (fig. 13) (Sirbu et al., 2005).
In zebrafish the cyp26 genes show dynamic expression patterns that progressively exclude RA from
the hindbrain as well (Hernandez et al., 2007).
18
Studies do suggest some redundancy between the function of the Cyp26 genes in the hindbrain.
Hindbrain patterning of Cyp26c1 and Cyp26b1 mutant mice was normal and Cyp26a1 mutants only
show a mild posteriorization (reviewed by Rhinn et al., 2012). A compound mutation of Cyp26a-/Cyp26c-/- however leads to strong posteriorization and loss of segmentation (reviewed by Rhinn et
al., 2012). Similarly, in zebrafish the subtle hindbrain patterning defects of the cyp26a1 mutant
became progressively more severe upon knock-down of cyp26c1 alone or knock-down of both
cyp26c1 and cyp26b1 (Hernandez et al., 2007), whereas knock-down of cyp26c1 and cyp26b1 did
not lead to patterning defects of the hindbrain, aside from a slight shortening of the hindbrain.
Fig. 13 Dynamically expressed Cyp26 enzymes shift the anterior limit of RA gradient posteriorly over time.
Dynamic expression of Cyp26 family members limits the extent of the RA gradient. In mice, RA activity is first detected
up to the r2/r3 rhombomere boundary. At this time Cyp26a1 is expressed in the anterior rhombomeres. Later, Cyp26c1
expression becomes expressed in r4 and the RA gradient regresses up to the r4/r5 boundary. From E8 on Cyp26b1 starts
to be expressed in r3 and r5, however RA activity (or more precisely: galactosidase activity) is still present in r5 and
further posteriorly at E8.5. Possibly the Cyp26b1 activity at this time is not sufficient to clear all RA from this region, or
Cyp26b1 does not have large effect at this time. Cyp26b1 mutant mice do not display any hindbrain defects. Also knockdown of cyp26b1 in the cyp26a1-/- background has only slight effect in zebrafish, which is much weaker than the effect
of cyp26c1 knockdown in this background. Adapted from: Sirbu et al., 2005
Thus, the domain of RA activity is established by diffusion of RA from the somites into the
hindbrain in anterior direction, forming a gradient, and is restricted at the anterior end by catabolism
of RA by CYP26 enzymes. First its activity is only excluded from the anterior hindbrain and later
also from more posterior rhombomeres, as a result of changing domains of CYP26 enzyme activity.
However, it cannot be excluded that regulation of RA receptor expression may also play a role in
controlling RA signaling. RAR expression in the hindbrain is dynamic and changes during
development (Hale et al., 2006; Mollard et al., 2000; Serpente et al., 2005), thus regulation of RA
receptor presence may also be a way to regulate RA signaling.
19
The RA gradient in the hindbrain specifies rhombomere identity in the posterior hindbrain
RA synthesized in the somites diffuses into the posterior hindbrain, forming a posterior (high) to
anterior (low) gradient in the hindbrain. RA posteriorizes the hindbrain in a concentration (and time)
dependent manner (fig. 14), presumably through sequential activation of the Hox genes (see next
section). Thereby it provides positional identity to the future rhombomeres r3-r8. This
posteriorization is needed for correct, rhombomere specific gene expression in the rhombomeres and
thus for correct specification of the rhombomeres. Expression of Hox genes, Krox20, Kreisler, vHnf1
and Fgf3 in the hindbrain are required for development of the rhombomeres. Indirectly, correct RA
signaling is also required for rhombomeric segmentation; the rhombomere specific expression of the
Eph receptors needed for segment boundary formation is lost in Raldh2 mutants (Niederreither et al.,
2000).
Anteriorly, CYP26 enzymes catabolize RA, thereby limiting the extent of RA activity and protecting
the anterior hindbrain from the posteriorizing effect of RA. GBX2 and FGF8, whose genes are
expressed in the anterior hindbrain, promote specification of the anterior rhombomeres, and OTX2
expressed in the future mid- and forebrain promotes mid- and forebrain development. Gbx2 is
required for anterior hindbrain development (of r1-r3) (Burroughs-Garcia et al., 2011). Furthermore,
GBX2 and OTX2 together position the midbrain-hindbrain boundary and the isthmus located here
(an organizer); the boundary arises at the border between their two expression domains (BurroughsGarcia et al., 2011; Sunmonu et al., 2011; Rhinn and Brand, 2001). Fgf8 is expressed on the
hindbrain side of the mid-hindbrain boundary, while Wnt1 is expressed on the midbrain side of the
border (Liu and Joyner, 2001). Initially their expression domains are quite broad, with Fgf8 being
expressed in the entire r1 and Wnt1 in the midbrain, but they become restricted to two narrow bands
on either side of the mid-hindbrain boundary (Liu and Joyner, 2001). Eventually, the Fgf8 expression
domain becomes restricted to the isthmus. The signals emanating from the isthmus help pattern both
the anterior hindbrain and posterior midbrain (Sunmonu et al., 2011) and FGF8 is the key molecule
for the function of this organizer (Sunmonu et al., 2011).
After segmentation of the hindbrain, RA induces Hoxb5-Hoxb8 gene expression in r7/r8 of the
posterior hindbrain (Oosterveen et al., 2003) (the authors did not specify the anterior limit of the
expression domains of each gene individually, although their anterior boundaries do seem to differ).
These two rhombomeres are less well-defined. This induction of Hoxb5-Hoxb8 might be required for
the specification of the identity these two rhombomeres.
20
Fig. 14 Retinoic acid establishes A-P patterning of the posterior hindbrain in a concentration dependent manner.
This is a schematic overview of the role of RA in patterning of the hindbrain. Rhombomere specific gene expression
patterns are depicted as they are observed around E8.25-E8.5, prior to hindbrain segmentation. Keep in mind however
that both the extent of the RA gradient, the RA levels and the expression domains of Cyp26 family members, Gbx2 and
Fgf8 are dynamic and change during patterning and development of the hindbrain. This is also the case for part of the
marker genes mentioned here. Kreisler expression, for example, is dynamic as well. In chick Kreisler is expressed in the
prospective r5 and r6 from the 5 somite stage onward, but it extends into r7 and r8 between somite stage 6 and 10
(Giudicelli et al., 2003). In zebrafish Fgf8 is additionally expressed in r4.
Inhibitory actions are depicted with long lines ending with a bar ( ---| ), while stimulatory actions are depicted with long
arrows. The small arrows indicate that FGF8 diffuses from the anterior hindbrain into the posterior midbrain.
*: Oosterveen et al. did not specify the anterior limit of the expression domains of each Hox gene individually, although
their anterior boundaries do seem to differ. Marker gene expression is based on the publications of Niederreither et al.
2000, Gavalas and Krumlauf 2000 and Kim et al., 2005.
RA activates Hox gene expression in the hindbrain in a sequential manner
As neural tissue is laid down by the regressing node, Hox gene expression is initiated in the newly
formed tissue. Only after the neural tissue is laid down by the node, these Hox genes become
sensitive to RA. In response to RA, the expression domains of the anterior and central Hox genes
(Hoxb1-Hoxb8) expand anteriorly so that the anterior limit of their expression reaches its definitive,
more anterior location in the posterior hindbrain (mouse) (Gould et al., 1998; Oosterveen et al.,
2003; Deschamps and Van Nes, 2005; Young and Deschamps, 2009). This anterior expansion is
dependent on RAREs located within the Hox clusters and this anterior expansion happens
exclusively in the neural tube, not in the mesoderm (Oosterveen et al., 2009; Deschamps and Van
21
Nes, 2005). The Hox genes become sensitive in a sequential order (3’ to 5’) with the first genes to
shift before E8.5 and the more 5’ Hoxb8 to start shifting at E10.5 (Gould et al., 1998; Oosterveen et
al.; reviewed by Deschamps and Van Nes, 2005). The anterior-most boundary of the Hox genes of
paralogous group 4, and other 3’ genes, are already met before hindbrain segmentation (Gould et al.,
1998; Niederreither et al., 2000). Additional regulatory inputs from rhombomere specific
transcription factors, such as KROX20 (EGR2), kreisler (MAFb/VAL), vHNF1 (HNF1b) and autoand cross-regulatory loops between Hox genes themselves, modulates Hox gene expression as well
(reviewed by Deschamps and Van Nes, 2005 and Glover et al., 2006; Wong et al., 2011). This way
the early Hox gene expression patterns in the hindbrain are established.
The anterior-most boundary of the Hoxb5-Hoxb8 genes is only reached after rhombomere
segmentation (Oosterveen et al., 2003). Hoxb5 becomes sensitive to RA at E9, Hoxb6 at E9.5 and
Hoxb8 at E10.5. By E11.5 their definitive boundaries have been reached. Oosterveen et al. proposed
that the expansion of Hox5-Hox8 gene expression into the hindbrain might contribute to patterning of
r7 and r8.
Initial anterior-posterior identity in the hindbrain might be provided by RA induced expression of
Hox genes
As transcription factors such as Krox20 and kreisler are expressed in rhombomere specific
expression domains, the question is what provides the hindbrain cells with their initial positional
identity along this axis to define these rhombomere patterns. A continuous gradient has to be
translated into discrete segmental (i.e. rhombomere specific) expression patterns. One theory is that
this initial anterior-posterior identity is provided by sequential, collinear 3’ to 5’ Hox gene
expression (Deschamps and Van Nes, 2005), but only for the caudal rhombomeres r3-r8, as Hox
gene expression is prevented in r1-r2. This could be a plausible theory, based on the dynamic Hox
gene expression patterns in the hindbrain.
The Hox clusters contain RA responsive elements and can therefore be directly regulated by RA. The
anterior expansion of the expression domain of the first Hox genes expression precedes the onset of
expression of Krox20 in r3 and r5 and the restriction of Fgf32 to r5-r6 (Giudicelli et al., 2001; Makki
and Capecchi, 2011; Vendrell et al., 2013). In chick, the anterior expansion also precedes the
expression of kreisler (Aragon and Pujades, 2009), but in mouse it occurs either just before or around
the onset of expression of kreisler (Cordes and Barsh, 1994; Kim et al., 2005).
Furthermore, Hox genes seem to be involved in the initiation of expression of at least some
rhombomere specific genes or in the restriction of expression of such genes to specific rhombomeres.
Hox genes are for example involved in initiating Krox20 expression in r3 and r5. In r3 the Hox genes
and MEIS2 together activate Krox20 and experiments suggest that HOXA1 might indirectly activate
Krox20 in r5 through activation of vHnf1 (Hnf1b) (Wassef et al., 2008; Makki and Capecchi, 2011).
Experiments indicate that the expression of genes such as Fgf3 and kreisler might be regulated by
Hox genes as well (Wassef et al., 2008; Makki and Capecchi, 2011; Pasqualetti et al., 2001).
However, many more regulatory actions are going on at the same time, and RA is also able to target
many other genes, thus it is possible that initial anterior-posterior identity is conferred by more genes
than only the Hox genes.
2
Fgf3 is first expressed throughout the rhombencephalon and later it becomes restricted to r5-r6 (Vendrell et al., 2013)
22
Establishment of the collinear expression of the Hox genes in the hindbrain seems to be based on
differential sensitivity of the Hox genes to RA. Several studies indicated that the 3’ Hox genes are
most sensitive to RA (induced at low concentrations of RA), whereas subsequent Hox genes in the
cluster are less sensitive and thus require higher concentrations of RA to become activated (reviewed
by Deschamps and Van Nes, 2005). RA forms a concentration gradient as it diffuses from the
somites into hindbrain; the differential sensitivity to RA then results in a specific Hox gene
expression pattern along A-P axis. 3’ Hox genes become expressed most anteriorly, while the more
5’ genes in the cluster are expressed at progressively more posterior levels. This Hox gene
expression in the hindbrain provides it with its positional identity. Further regulatory inputs from
rhombomere specific transcription factors and auto- and cross-regulatory loops between Hox genes
themselves, then modulate the Hox gene expression patterns to establish their rhombomere specific
expression patterns in the hindbrain (reviewed by Deschamps and Van Nes, 2005 and Glover et al.,
2006; Wong et al., 2011). For example, Hoxb1 is first expressed in a broad region in the hindbrain
(and spinal cord) and only later it becomes restricted to a small band of tissue of the future
rhombomere r4 (it remains expressed in spinal cord) (Wong et al., 2011; Glover et al., 2006).
Examples of compounds affecting neural tube development
Ethanol
One compound that is very well-known for its effect on brain development and the adult brain, is
ethanol, commonly known as alcohol. During ethanol detoxification ethanol is first converted to
acetaldehyde, which is very toxic, and then to acetic acid. Alcohol abuse during pregnancy can result
in a wide range of defects in the unborn child, referred to as Fetal Alcohol Spectrum Disorder
(FASD). Exposure to EtOH can result in embryonic lethality, stillbirth, or developmental defects
(Kot-Leibovich and Fainsod, 2009). Children with FAS can have, amongst other defects, craniofacial
malformations, microcephaly, and microphthalmia. These children experience neurological defects.
Various studies have implicated that deregulation of RA signaling by EtOH may at least in part
account for the toxic effects of EtOH (Deltour et al., 1996; McCaffery et al., 2004; Yelin. 2005;
Yelin 2007; Kot-Leibovich and Fainsod, 2009; Kumar et al., 2010; Kane et al., 2010). First of all,
FAS and the developmental phenotypes caused by ethanol exposure resemble the phenotype of
embryos with reduced RA signaling as well as to those with excess RA (Yelin. 2005). Second, in
Xenopus and zebrafish embryos the defects caused by ethanol can be partially rescued by
supplementation with retinoids (Yelin et al., 2005; Marrs et al., 2010). In Xenopus embryos treatment
with EtOH at the gastrula stages leads to impaired head development, with a reduced forebrain,
shortening along the rostro-caudal axis and microphthalmia (reduced eye) (Yelin et al., 2007).
Raldh2-/- mouse embryos for example show severe impaired development as well and die
midgestation, they display amongst other defects shortening along the A-P axis, incomplete closure
of the neural tube and reduction of frontonasal region (Niederreither et al., 1999). Also at later stages
of development there are similarities between the effects of EtOH exposure and disturbed RA
23
signaling. In the third trimester of humans or murine postnatal days 4-9 the cerebellum is especially
sensitive to both EtOH exposure and disturbances in RA signaling and their effects on the
cerebellum show similarities (McCaffery et al., 2004; Kumar et al., 2010). Experiments suggest that
at least part of the toxic effects of EtOH may be the result of altered RA signaling. The effects of
ethanol on RA signaling, however, are very much dependent on developmental stage (and tissue).
The effects of EtOH on early neural development will be discussed in detail in the next section. After
that, the effects of EtOH on brain development during later stages are briefly discussed as well.
Ethanol competes for RALDH2 activity during gastrulation stages
Experiments suggest that at least part of the abnormalities observed after prenatal alcohol exposure
are the result of abnormally low RA levels due to competition with retinaldehyde for RALDH2
activity by ethanol (Deltour et al., 1996; Kot-Leibovich and Fainsod, 2009). First of all, FAS and the
developmental phenotypes caused by ethanol exposure during embryonic development resemble
those of embryos with reduced RA signaling as well as to those with excess RA (Yelin. 2005).
Ethanol treated embryos display impaired head development, with a reduced forebrain size,
shortening along the rostro-caudal axis, microphthalmia (reduced eye) and hindbrain defects (Yelin
et al., 2007; Marrs et al., 2010). Raldh2-/- mouse embryos for example show severe impaired
development and die midgestation, they display a.o. shortening along the A-P axis, incomplete
closure of the neural tube and reduction of frontonasal region (Niederreither et al., 1999). Second,
RA reporter levels were strongly decreased in embryos exposed to ethanol around gastrulation stages
in both Xenopus and mouse (Kot-Leibovich and Fainsod, 2009; Deltour et al., 1996). In Xenopus it
was demonstrated that the expression of RA responsive genes Hoxb1 and Hoxb4 was reduced as well
(Kot-Leibovich and Fainsod, 2009). Expression of Cyp26a1, which is involved RA degradation
expression and can be regulated by RA, was also reduced.
In support of the notion that part of the defects of ethanol exposure are due to lower RA levels, is the
fact that retinoid supplementation was able to partially rescue the effects ethanol exposure in
zebrafish and Xenopus (Yelin et al., 2005; Marrs et al., 2010). It partially rescued the impaired
gastrulation movements and the concomitant shortening of the axis, hindbrain development and
craniofacial defects (Yelin et al., 2005; Marrs et al., 2010).
It seems that the effect of EtOH on RA signaling is through competition for RALDH2 activity (KotLeibovich and Fainsod, 2009). Suppression of RALDH2 activity in combination with ethanol
treatment aggravated the reduction of RA signaling, the reduction of expression of RA responsive
genes, as well as the phenotype. On the other hand, overexpression of Raldh2 in part rescued the
phenotype, RA signaling and RA responsive gene expression. Furthermore, it seems that EtOH acts
mostly through competition for RALDH2, and no other enzymes, as EtOH treatment did not
aggravate the phenotype of Raldh2 knock-down embryos, which would be expected if EtOH also
targets other enzymes. The embryos are most sensitive to EtOH exposure at the onset of RA
signaling (Yelin et al.,2005; Kot-Leibovich and Fainsod, 2009), when RA levels are still low, and the
availability of RALDH2 is also relatively low at this point. The defects are also cumulative: defects
are more severe upon longer exposure. Exposure of embryos at the neurula stages (stage 18 and
onward) did not reveal any gross morphological defects in the over-all embryo (Yelin et al., 2005),
however patterning of the embryo was not examined in detail.
24
In normal Xenopus development, RA negatively regulates expression of organizer specific genes. In
alcohol treated embryos the expression of organizer-specific genes was upregulated or expanded,
indicating that the negative control by RA is lost. Again, overexpression of Raldh2 seems to rescue
the expression levels of these genes (Kot-Leibovich and Fainsod, 2009). Experiments suggest that
most of the defects observed arise because of disruption of the organizer. The rostral-caudal
shortening seems to be mediated through impaired convergence extension-movements, which may
be caused by the expansion of the expression of Otx2 in the organizer upon EtOH exposure (Yelin et
al., 2005).
Thus it seems that at least part of the teratogenic effects ethanol are mediated by its competition for
RALDH2 activity with retinaldehyde (fig. 15). As a result RA levels decrease upon ethanol
exposure, which leads to loss of negative regulation of organizer specific genes and defects that
resemble the RA deficiency phenotype. In the light of the previous chapters, we see again that RA
seems to function to antagonize signals from the organizer; the node. It would be interesting to test if
differentiation and rostro-caudal patterning are affected as well, as loss of RA signaling leads to an
expansion of Fgf8 expression domains, impaired neural differentiation, reduced number of neurons
in the spinal cord and anteriorization of the hindbrain with loss of rhombomere segmentation. Also,
both ethanol and aberrant RA levels can cause malformations of the hindbrain (McCaffery et al.,
2004). This could be tested by studying the effect of EtOH on the expression levels and expression
patterns of Fgf8, Wnt3a, the Hox genes, and hindbrain patterning genes - such as Krox20, kreisler,
and vHnf1 – and of neural differentiation genes, such as Ngn1,Ngn2, Neurod4. Interestingly, Pax6
expression in the hindbrain is reduced upon EtOH exposure (Santos-Ledo et al., 2013), but
unaffected in the anterior structures, this may be an indication that hindbrain patterning is affected
(Kayam et al., 2013).
A role for RA signaling in forebrain development or in patterning of the surrounding mesoderm may
account for the reduction of the forebrain and eye in ethanol treated embryos. Raldh2 is transiently
expressed in the anterior neural plate and optic vesicles and later Raldh3 is expressed in the
overlying ectoderm (reviewed by Rhinn and Dollé, 2012). Interference with RA signaling, by knockout of Raldh2 or dominant negative RA receptors, might interfere with patterning of the forebrain,
and it resulted in decreased cell proliferation and survival in the forebrain. At later stages, RA may
be involved in the development of the cortex of the forebrain (Rhinn and Dollé, 2012; Kane et al.,
2010). The exact effects of ethanol on RA signaling, however, is dependent on stage, and tissue, thus
a later effect of ethanol on forebrain development, may not be through downregulation of RA, but
through excess RA or altering of RA signaling (see next sections). Lack of RA signaling also leads to
smaller somites (Rhinn et al., 2012), thus smaller somites might also be expected in EtOH exposed
embryos.
25
Fig. 15 Proposed adverse outcome pathway of embryonic EtOH exposure during gastrulation stages
Ethanol (EtOH) exposure during embryonic development can lead to developmental abnormalities, such as shortening
along the anterior-posterior axis, hindbrain defects, impaired head development with a reduced forebrain and
microphthalmia. In the developing embryo EtOH leads to expansion of the domains of organizer specific genes. The
associated disruption of the organizer then leads to impaired convergence-extension movements resulting in a shortening
along the anterior-posterior (A-P) axis. Ethanol competes with retinol for RALDH2 activity. As a result RA synthesis is
reduced, resulting in subnormal RA levels. A potential mechanism which may explain part of the teratogenic effects
upon EtOH exposure was extrapolated, based on the knowledge about the roles of RA in patterning of the embryo, the
considerable overlap between defects observed upon EtOH treatment and RA depletion, the phenotypes observed in
EtOH treated embryos and the rescuing effect of Raldh2 overexpression or retinoid supplementation.
Ethanol exposure changes RA levels in the hippocampus and cortex during late embryonic
development as well as in the adult brain
The hippocampus is sensitive to both EtOH and aberrant RA levels. Prenatal EtOH exposure leads to
impaired formation of dendritic spines in the hippocampus and abnormal development (Kane et al.,
2010). In contrast to the effects of EtOH during early development, EtOH exposure between E13 and
E19 strongly increased RA levels in the hippocampus and cortex of E19 mouse embryos, as well as
retinol levels (Kane et al., 2010). RA and retinol levels were also increased in the adult hippocampus
and cortex upon chronic EtOH exposure. The experiments in adult rats suggest that the increase of
RA levels is likely the result of both increased uptake of retinol and RA from the blood stream and
increased RA synthesis due to increased RALDH1 activity3, aided by a modest increase in RDH
3
Not Raldh1 expression; RALDH1 protein levels were decreased, whereas RALDH1 activity was increased.
26
activity. In the hippocampus RA is involved in neurogenesis and normal dendritic spine formation
and branching (Kane et al., 2010). Thus EtOH treatment of E13-E19 rat embryos results in increased
RA levels in the cortex and hippocampus. This strong increase of RA levels may impair neuronal
cell division and affect the formation of dendritic spines and may thereby contribute to the defects
associated with fetal alcohol syndrome.
Ethanol exposure results in increased RA synthesis and altered RA signaling in the developing
cerebellum
In rats ethanol exposure inhibits differentiation and causes increased cell death in the developing
cerebellum. In humans cerebellar development takes place in the third trimester of pregnancy, in rats
this occurs at postnatal days 4-9. The cerebellum is one of the brain areas most sensitive to RA and
EtOH (McCaffery et al., 2004; Kumar et al., 2010). Ethanol treatment increases RA levels in the
cerebellum (McCaffery et al., 2004), presumably by increasing RA synthesis by astrocytes.
McCaffery et al. suggested that EtOH does so by stimulating shortchain retinol dehydrogenases, but
this was not tested. Furthermore, ethanol exposure in rats results in altered RAR and RXR receptor
expression and activity (Kumer et al., 2010), which may result in altered responses to RA. The
increase in RXR activity may account for the increased apoptosis, as RXR targets apoptosis related
genes (Kumar et al., 2010). RAR activity on the other hand was decreased, which may account for
the decreased neural differentiation upon EtOH treatment. In adult mice, EtOH did not affect
cerebellar RA levels anymore (Kane et al., 2010).Thus the toxic effects of EtOH on cerebellar
development may be mediated by altered RA levels and signaling.
The effect of Ethanol exposure on RA signaling is stage dependent
Thus to summarize, there is considerable overlap between the teratogenic effects of EtOH and
disturbed RA signaling on neural development and it seems that at least a part of the teratogenic
effects of EtOH on neural development can be contributed to the effect of EtOH on RA signaling.
The specific effect of ethanol on RA signaling is very much dependent on developmental stage (and
tissue). Early on, at the onset of RALDH2 activity when RALDH2 and RA levels are still low,
ethanol competes for RALDH2 activity and this leads to a reduction of RA levels, which may
account for at least some of the teratogenic effects of EtOH during early development. Later, EtOH
may increase the activity and expression levels of RA synthesizing enzymes and the expression of
enzymes involved in retinol uptake in specific brain areas, resulting in higher retinol and RA
concentrations in these tissues. This combined results in a drastic increase of RA levels which may
account for the observed late developmental defects observed resulting from EtOH exposure.
Furthermore, EtOH may alter the response to RA through its effect on RAR and RXR receptor
expression (Kumar et al., 2010).
27
Triazoles
Triazoles are used as antifungal agents in agriculture and medicine and affect fungal cell wall
integrity through inhibition of the CYP51 enzyme (Robinson et al., 2012). Examples of triazoles are
flusilazole (FLU), cyproconazole (CYP) and triadimefon (TDI). These triazoles can have teratogenic
effects on development, including neural tube development. Defects include axial defects,
craniofacial defects, and defects in hindbrain patterning and segmentation. Skeletal analysis revealed
axial abnormalities, including transformation, fusion or duplication of axial segments, suggesting
that the specification of axial identity is affected by triazoles (Menegola et al., 2005a). As the
establishment of axial patterning in the mesoderm and neural ectoderm involves similar mechanisms,
it seems likely that neural patterning is affected as well. Exposure to triazoles at the beginning of
somitogenesis also affects rhombomere patterning; KROX20 protein expression in the murine
hindbrain was reduced and seemed scattered in triazole treated embryos (Menegola et al., 2004;
Menegola et al., 2005a; Menegola et al., 2005b). In the in vitro rat system HOXB1 protein
expression was reduced and scattered as well (Menegola et al., 2004). This scattering could be due to
impaired boundary formation in the rhombomeres: correct rhombomere specific, alternating Eph
expression is needed to prevent cell mixing between rhombomeres. The experiments with the rat in
vivo and mouse in vitro system suggested that the craniofacial defects of triazole treated embryos
may be the result of altered neural crest migration patterns, resulting from the abnormal rhombomere
patterning.
Triazoles interfere with RA signaling
Several studies suggest triazoles might interfere with RA levels. This is based on the similarities
between phenotypes of RA and triazole treated embryos, the changes of expression of genes
involved in RA metabolism upon triazole exposure and the substantial overlap between gene
expression changes upon treatment with Flusilazole or RA in whole embryo cultures (WEC)
(Menegola et al., 2004; Menegola et al., 2005a; Robinson et al., 2012).
First of all, RA is known to be involved in patterning along the anterior-posterior axis in both
mesoderm and neural ectoderm and the axial defects of TDI treated embryos were similar to those
observed in RA treated embryos (Menegola et al., 2005a). Also the effect of RA or triazole treatment
on hindbrain patterning was similar, and the effects were additive as treatment with subteratogenic
doses of both RA and Fluconazole led to the same phenotype as the teratogenic dose of Fluconazole
alone (Menegola et al., 2004).
Second, genes involved in RA metabolism were upregulated in response to triazole treatment and
triazoles altered the expression of genes involved in hindbrain patterning, a process which is
dependent on and regulated by RA. Cyp26a1 was strongly upregulated in the rat WEC and in
zebrafish, as well as Dhrs and Crabp2 (Robinson et al., 2012; Hermsen et al., 2012). In zebrafish the
expression of the major gene involved in RA synthesis in early development, Raldh2, was
downregulated, while genes involved in storage and degradation of RA were upregulated (Hermsen
et al., 2012). Hindbrain patterning genes Krox20 (Egr2) and kreisler (Mafb) were downregulated in
the rat WEC, whereas Gbx2 is upregulated (Robinson et al., 2012). Disturbed patterning, with a
reduction of Krox20 expression was already shown before in vivo and in vitro. Some Hox genes and
vHnf1 (Hnf1b) showed altered expression as well in zebrafish embryos (Hermsen et al., 2012). An
28
upregulation of Gbx2 may seem contradictory with an upregulation of RA, as Gbx2 is involved in
patterning of the anterior rhombomeres while (excess) RA has a posteriorizing effect the hindbrain.
However, Gbx2 expression in the hindbrain is dynamic, and Gbx2 expression is not limited to the
hindbrain; it is also expressed in the posterior embryo (Martinez-Barbera et al., 2001). During
headfold to presomite stages Gbx2 is broadly expressed in a large part of the embryo, later it
becomes more restricted to the caudal and hindbrain areas. Because of this, the change of Gbx2
expression may not be very telling; it does not tell in which part of the embryo its expression is
affected, and if there is a (slight) delay in development due to the treatment4, higher levels of Gbx2
may be observed as well (as it is still expressed in a larger domain).
Third, the sets of differentially expressed (DE) genes between RA and triazole treated whole
embryos showed significant overlap (31 of the 62 DE genes in triazole treated embryos were also DE
in RA treated embryos), and these genes showed a similar response to either of the treatments in
terms of expression changes of these genes (Robinson et al., 2012). Thus, the effects of triazole
exposure show similarities to the effects of RA excess, on both the molecular and phenotypic level.
Additionally, triazole exposure affects the expression of at least some genes involved in RA
metabolism. This suggests that triazoles may elicit their teratogenic effects at least in part through
deregulation of RA levels.
Triazoles inhibit CYP26 activity
Triazoles may deregulate RA levels by inhibiting of CYP26A1 (Robinson et al., 2012). Experiments
indicate triazoles might interact with the active site of CYP26A1 (Ren et al., 2008; Thatcher et al.,
2011; Gomaa et al., 2012). However if teratogenic effects of triazoles are caused by the inhibition of
CYP26 enzymes, it seems more likely that triazoles cause these effects by incompletely inhibiting
the activity of multiple CYP26 members as knock-out of Cyp26a1 leads to much more severe axial
defects than observed in triazole treated embryos, but its complete deletion only results in a mild
hindbrain phenotype and a relatively normal craniofacial phenotype. Cyp26a1 deletion causes severe
body axis truncation, well before the caudal levels (Sakai et al., 2001; Abu-Abed et al., 2001),
whereas triazole treated embryos did develop caudal vertebrae. This suggests that CYP26A1
inhibition is not complete. Cyp26a1 deletion did cause an anterior transformation of the first lumbar
vertebra to a thirteenth thoracic vertebra identity (Sakai et al., 2001), thus the anterior
transformations and other segmental phenotypes in triazole treated embryos could in theory be the
result of CYP26A1 inhibition. On the cervical levels, however, Cyp26a1 deletion caused posterior
transformations (Sakai et al., 2001; Abu-Abed et al., 2001). The effect of complete genetic ablation
of Cyp26a1 on hindbrain patterning however is very mild, with only a slight effect on Krox20
expression patterns (Sakai et al.,2001; Abu-Abed et al., 2001). There also was a relatively normal
craniofacial appearance (Abu-Abed et al., 2001). Compound knock-out of Cyp26a1 and Cyp26c1
does lead to severe, more widespread hindbrain patterning defects, failed neural crest migration and
stronger craniofacial defects (Uehara et al., 2007), but it is embryonic lethal in mice before
embryonic day E11. Cyp26b1 single deletion is also associated with abnormal neural crest migration
and craniofacial defects, but does not seem to alter hindbrain patterning (Maclean et al., 2009). It
4
Triazole treatment does seem able to delay development. TMS scores as well as somite count are lower in triazole
treated whole rat culture embryos 48 hours after a single dose exposure (Robinson et al., 2012), and in triazole treated
zebrafish embryos development is delayed as well(Hermsen et al., 2012)
29
thus seems likely that other CYP26 members are partially inhibited as well. The idea that the
inhibition of multiple Cyp26 enzymes results in more widespread defects in the hindbrain is in line
with the earlier proposed model in which dynamic Cyp26 expression regulates the extent of the RA
gradient in the hindbrain, with some functional redundancy between the Cyp26 members.
The strong upregulation of Cyp26a1 expression in triazole treated embryos (rat whole embryo
culture / zebrafish), is also seen in embryos exposed to excess RA and is likely part of a feedback
mechanism to regulate RA levels.
A potential mechanism for RA mediated teratogenic effects of triazoles
Considering the effects of RA and triazole treatment on patterning of the embryo, phenotype and
gene expression a potential mechanism for RA mediated effects of triazoles on development of the
neural tube could be the following (fig. 16):
Triazoles partially inhibit CYP26A1, and likely the other CYP26 members as well, which then
results in locally increased, ectopic levels of RA. This will especially affect the caudal region
containing the posterior growth zone and the hindbrain. In the posterior growth zone decreased
CYP26A1 activity leads to ectopic RA in this region. RA may then negatively affect FGF8 levels in
this region. As increasing Fgf8 levels are needed for specification of increasingly posterior tissues in
the spinal cord, this lowering of FGF8 levels may lead to faulty specification of axial identity
(segment identity) and thus to disturbed patterning along the A-P axis. More specifically, it may lead
to altered segment identity such as an anterior transformation. Lower FGF8 levels in and around the
node induce a less posterior identity in the cells leaving this area than is normal at the particular axial
level. This is reflected by an altered induction of Hox gene expression. Proliferation rates might be
decreased as well in the posterior growth zone due to lower levels of FGF8. In the posterior
hindbrain a decrease of CYP26 expression may result in higher and ectopic RA levels in the
hindbrain. This may lead to faulty Hox gene expression patterns and to faulty rhombomere
specification and identity. In turn this leads to altered hindbrain development and disturbed neural
crest migration patterns. The latter could then result in craniofacial defects. In addition, disturbed RA
signaling in the branchial arches may contribute to the craniofacial deformities as well, as Cyp26
enzymes are expressed in the branchial arches (Rhinn and Dollé, 2012).
30
Fig. 16 Proposed adverse outcome pathway of embryonic triazole exposure, focusing on the effects on neural tube
patterning and the embryonic axis.
Triazole exposure during embryonic development can lead to developmental abnormalities, such as shortening along the
anterior-posterior (A-P) axis, patterning defects along this axis, hindbrain defects and craniofacial defects. Triazoles were
shown to affect the expression of some hindbrain patterning genes (e.g. Krox20, Hoxb1). Studies suggest that these
hindbrain patterning defects result in disturbed neural crest cell migration patterns and that this may account for the
craniofacial defects. However Cyp26 expression is also observed in the branchial arch mesenchyme, thus increased or
ectopic RA levels in the branchial arches may contribute as well to these defects. Triazoles are able to inhibit CYP26A.
The resulting ectopic or increased RA levels may account for part of the observed developmental defects. A potential
mechanism which may explain part of the teratogenic effects of triazole exposure was extrapolated, based on the
observed developmental effects in triazole treated embryos, the current knowledge of RA function in the developing
embryo, the considerable overlap between effects of triazole and RA treatment on development and gene expression, and
the similarities with the phenotypes exhibited by Cyp26 deficient embryos.
In addition to disruption of axial and hindbrain development, inhibition of CYP26 enzymes could additionally result in
higher RA levels in the growth buds of other developing organs, such as the limb buds, resulting in developmental
defects there. CYP26 enzymes are for example also expressed in the limb buds and branchial arch mesenchyme
(reviewed by Rhinn and Dollé, 2012) and triazoles are known to affect development of these structures (Robinson et al.,
2012).
31
With this model in mind, interesting genes to look at in relation to assessment of potential neurotoxic
effects would be:
Fgf8 – this gene is involved in axis elongation and patterning, excess RA can downregulate
expression of this gene. Fgf8 is also interesting because it is expressed in the growth buds of
several developing organs in which RA plays a role as well. In the case of triazoles a decrease
in Fgf8 may be expected. Wnt3a might also be interesting in this respect.
Hox genes – these genes confer positional identity to the spinal cord and posterior hindbrain.
If FGF8 levels are lower in the posterior growth zone, it might be expected that the posterior
Hox genes are induced less or only at more posterior levels than normal in the developing
spinal cord. The more 3’ Hox genes would be induced in more posterior tissues than normal.
In the hindbrain the Hox genes are involved in patterning and segment identity. Increased RA
levels might result in an enhanced anterior expansion of the expression domains of the Hox1Hox5 genes, i.e. their domains expanding more anteriorly than normal, but since there are a
lot of feedback loops involved in establishing their rhombomere specific expression domains,
the effect of RA on the Hox expression patterns at the rhombomere specification stages might
be hard to predict.
The hindbrain patterning genes described previously, such as Krox20 (Egr2), kreisler (Mafb),
Fgf3, Gbx2, Meis2, Otx2, vHnf (Hnf1b) – altered of expression of these genes reflect
disturbed anterior-posterior patterning of the hindbrain.
Neural differentiation genes, such as Neurogenin1 (Neurog1/Ngn1), Neurogenin2
(Neurog2/Ngn2), NeuroM (Neurod4) – the presence of RA and a drop in Fgf8 are needed for
neuronal differentiation. Shh and Gli2 might be interesting in this respect as well. The
presence of RA and the drop of Fgf8 levels are also needed for activation of Shh in the floor
plate and for activation of other dorsal-ventral patterning related genes. RA dependent
repression of Gli2 seems necessary for the cells to be able to respond to the Shh signals
(Ribes et al., 2009).
Additionally, studying the expression of the genes involved in RA synthesis (most importantly
Rdh10, Raldh2), catabolism (Cyp26 family), retinol uptake (Stra6) and RA signaling (RAR and RXR
receptor family) may provide additional insight into potential effects on RA levels and RA signaling
activity. Measuring RA levels could be insightful as well.
Unfortunately, I did not have a look at the expression data to see whether these genes did change
their expression upon triazole exposure in the whole rat embryo culture (WEC), zebrafish embryos,
embryonic stem cell test (ESTc) or neural embryonic stem cell test (ESTn). Some of the genes listed
above were mentioned in the articles describing the rat WEC and zebrafish tests; these were
mentioned in the overview of the effects of triazole treatment in the text above. Interestingly, Fgf8
was downregulated upon triazole exposure in the neural Embryonic Stem Cell test (ESTn), while
Rarb and Cyp26b1 expression was increased (Theunissen et al., 2012). Expression of Hoxb4, Hoxc4
and Hoxb5 was upregulated as well.
32
Conclusions
The classic way to test for possible teratogenic effects of compounds is by exposing model
organisms (animals) to a high dose of the compound to see if development is affected (Krewski et
al., 2007). Because of the high amount of test animals is needed, the costs (time and money) and the
limited insight it provides in the mechanisms underlying the toxic effects, there is a demand for
alternative approaches. In modern toxicity research novel approaches are being developed for
assessment of potential risks. One of these approaches is based on the idea of looking at the effects
of a compound on specific key pathways, of which its distortion may be indicative of an adverse
effect on (for example) embryonic development. If such a key pathway is altered upon exposure to a
compound, this might enable the researcher to predict the developmental outcome based on which
components of the pathway are deregulated. Also, it might become possible to reconstruct (part of)
the sequence of events that leads to the developmental defects after exposure: the adverse outcome
pathway (AOP). The RA signaling pathway was proposed as a candidate pathway to study in relation
to neural tube development, due to its central role in development and the association of disturbed
RA signaling with neural tube defects. During early neural tube development RA is especially
important for anterior-posterior patterning of the spinal cord and hindbrain, neural differentiation and
axis elongation. Furthermore, RA is required to allow cells to respond to ventralizing Shh signals.
In order to be able to construct an AOP based on the effects of a compound on RA signaling, the role
of RA signaling in neural tube development was reviewed. The information of how a compound
affects RA signaling and what the effects are on the genes downstream of RA in the hindbrain and
spinal cord, might allow the deduction of the sequential steps that may lead to a developmental
phenotype. The effect of a compound on (embryonic) development is very dependent on the stage(s)
at which the embryo is exposed, the duration of exposure and the dose. This review focused on early
neural tube development; exposure at later stages may have quite different effects.
Potential neurotoxic risks may be identified by studying the effect of a compound on RA levels and
on signaling downstream of RA. If RA levels cannot be measured directly, changes in the expression
of genes associated with RA signaling, may be indicative of disturbed RA signaling. Genes involved
in RA synthesis (most importantly Rdh10, Raldh2), catabolism (Cyp26 family), retinol uptake
(Stra6) and RA signaling (RAR and RXR receptor family) may provide insight into potential effects
on RA levels and RA signaling activity. The potential effects on neural tube development could be
characterized by looking at marker genes that act downstream of RA. Interesting genes to study in
relation to early spinal cord development are Fgf8, the Hox genes and neural differentiation genes
such as Ngn1, Ngn2, Neurod4, Shh and Gli2. Genes such as Mafb (kreisler), Egr2 (Krox20), Meis2,
Hnf1b, Fgf3, Gbx2, En2, Pax2, Otx2 and the Hox genes from paralogous group 1-4 (Hox1-Hox4)
provide information on the effects on patterning of the hindbrain.
Potential effects of exposure to a compound on patterning of the neural tube, neural differentiation or
axis elongation might be revealed by in situ hybridization with probes for these marker genes.
Alternatively, gene expression levels could be compared between treated and control embryos, for
example by Q-PCR or micro-array analysis. If whole embryos are used for gene expression level
comparisons, interpretation of the results should be done with care. Some of the genes considered in
33
this review are expressed at multiple sites in the embryo, as they are involved in different processes.
This may make it difficult to pinpoint in which part of the embryo the expression is affected.
Additionally, this approach may not be sensitive enough to pick up local changes, nor will it detect a
shift (e.g. an anterior or posterior shift) of an expression domain. Also the expression of many genes
is dynamic during the course of development. The expression domains of some of the hindbrain
patterning genes, for example, either expand or shrink between the onset of hindbrain specification
and the stage of hindbrain segmentation. If there is a slight delay in development, this may also result
in altered expression patterns in the embryo as well, while hindbrain specification might be
unaltered. It is therefore recommended to match embryos based on developmental stage, rather than
age.
With the neural embryonic stem cell test (ESTn), it may become possible to do an initial assessment
of the potential risks of many compounds at once in a relatively short time scale (Theunissen et al.,
2013). Analysis of expression changes of aforementioned marker genes may give an indication
whether RA signaling is affected, however the time and region specific context may be lost. Thus
tissue specific downstream effects of RA signaling might not be picked up. The ESTn might thus
reflect the effects of compound exposure on an embryo less well, but it may still give an indication
whether RA signaling may be affected. In line with this notion, is the observation that the gene
expression changes in the embryonic stem cell test (ESTc) upon flusilazole treatment only show a
mild correlation with the gene expression changes in flusilazole treated zebrafish embryos (Robinson
et al., 2012). However, gene expression changes in the cells do indicate that RA signaling is affected
by triazole exposure; expression levels of the RA signaling associated genes Rarb and Cyp26b1
increased after exposure to another triazole (cyproconazole) (Theunissen et al., 2012). Also the
expression levels of a few Hox genes, which can be directly regulated by RA, were upregulated as
well. On the other hand, Fgf8 expression, which in vivo is suppressed by RA, was downregulated
upon triazole exposure (Theunissen et al., 2012). These expression changes could indicate that RA
signaling is increased upon triazole exposure, which is concordant with the suggestion that triazoles
may increase RA signaling in vivo. Thus, it may be possible to detect changes in RA signaling with
the ESTn and such a change in RA signaling may be an indication that the compound might affect
neural tube development in embryos.
For two types of toxicants, ethanol and triazoles, potential AOPs were proposed that linked the
molecular initiating event (MIE) – interaction of the compound with RALDH2 or CYP26
respectively – to the observed developmental defects. The AOPs were based on the effects of the
toxicants on RA signaling. The effects that the deregulation of RA signaling may have had on
(neural tube) development was then extrapolated, based on the known roles of RA signaling
described in this review and the resemblance between the developmental defects associated with
exposure to the compound and deregulated RA signaling. This led to a proposal of a series of
potential intermediate steps that might link the MIE to the observed developmental defects on the
organ and organismal level. Additional morphological and molecular analyses (e.g. gene expression
analyses, in situ hybridization) are needed to test the validity of the AOPs and to refine them.
Thus in the case of EtOH and triazoles exposure it was possible to pose hypothesis (the AOP) on
how compound exposure might have led to the associated developmental defects, based on current
knowledge of the role of RA in neural tube development and axis patterning. These AOP models
might then be used to further investigate the mechanisms underlying triazole and ethanol exposure.
34
Gene expression measurements after triazole exposure were already done in the rat WEC at the 2-4
somite stages and in the neural embryonic stem cell test (Robinson et al., 2012; Theunissen et al.,
2012). A revisit of these data, focusing on the previously mentioned list of genes of interest, may
already provide additional information that might help test the validity of the proposed AOP and to
refine or adjust it. Gene expression was also measured in the zebrafish embryo, however this was
measured at 24 hpf (Hermsen et al., 2012), at which point axis elongation is (nearly) completed and
the hindbrain has already segmented.
Thus, the RA signaling pathway and the associated genes (biomarkers) summarized here may in the
future be studied to assess potential risks posed by exposure to a compound in relation to early neural
tube development and to help building AOP networks. Changes in expression of these genes are
indicative of defects in anterior-posterior patterning of the spinal cord and hindbrain, or of disruption
of differentiation or axis elongation (organizer function). In the case of the teratogenic triazoles, gene
expression levels after exposure were already measured in several model systems; reexamination of
these data could provide new insights into the mechanisms underlying the teratogenic effects of
triazole exposure.
35
Acknowledgements
I’d like to express my special thanks to Prof. Dr. Piersma for supervising me and providing me the
opportunity to write my thesis at the RIVM, and Dr. Ilse Tonk, for supervising me and giving me
feedback on previous versions of this thesis.
References
Ankley, G.T., Bennett, R.S., Erickson, R.J., Hoff, D.J., Hornung, M.W., Johnson, R.D., Mount, D.R., Nichols, J.W.,
Russom, C.L. & Schmieder, P.K. 2010, "Adverse outcome pathways: a conceptual framework to support
ecotoxicology research and risk assessment", Environmental Toxicology and Chemistry, vol. 29, no. 3, pp. 730-741.
Aragon, F. & Pujades, C. 2009, "FGF signaling controls caudal hindbrain specification through Ras-ERK1/2 pathway",
BMC developmental biology, vol. 9, pp. 61-213X-9-61.
Bel-Vialar, S., Itasaki, N. & Krumlauf, R. 2002, "Initiating Hox gene expression: in the early chick neural tube
differential sensitivity to FGF and RA signaling subdivides the HoxB genes in two distinct groups", Development,
vol. 129, no. 22, pp. 5103-5115.
Blentic, A., Gale, E. & Maden, M. 2003, "Retinoic acid signalling centres in the avian embryo identified by sites of
expression of synthesising and catabolising enzymes", Developmental dynamics : an official publication of the
American Association of Anatomists, vol. 227, no. 1, pp. 114-127.
Burroughs-Garcia, J., Sittaramane, V., Chandrasekhar, A. & Waters, S.T. 2011, "Evolutionarily conserved function of
Gbx2 in anterior hindbrain development", Developmental dynamics : an official publication of the American
Association of Anatomists, vol. 240, no. 4, pp. 828-838.
Cordes, S.P. & Barsh, G.S. 1994, "The mouse segmentation gene kr encodes a novel basic domain-leucine zipper
transcription factor", Cell, vol. 79, no. 6, pp. 1025-1034.
Dasen, J.S., Liu, J. & Jessell, T.M. 2003, "Motor neuron columnar fate imposed by sequential phases of Hox-c activity",
Nature, vol. 425, no. 6961, pp. 926-933.
Deltour, L., Ang, H.L. & Duester, G. 1996, "Ethanol inhibition of retinoic acid synthesis as a potential mechanism for
fetal alcohol syndrome", FASEB journal : official publication of the Federation of American Societies for
Experimental Biology, vol. 10, no. 9, pp. 1050-1057.
Deschamps, J. & van Nes, J. 2005, "Developmental regulation of the Hox genes during axial morphogenesis in the
mouse", Development, vol. 132, no. 13, pp. 2931-2942.
Dias, M.S. & Partington, M. 2004, "Embryology of myelomeningocele and anencephaly", Neurosurgical focus, vol. 16,
no. 2, pp. 1-16.
Diez del Corral, R., Olivera-Martinez, I., Goriely, A., Gale, E., Maden, M. & Storey, K. 2003, "Opposing FGF and
retinoid pathways control ventral neural pattern, neuronal differentiation, and segmentation during body axis
extension", Neuron, vol. 40, no. 1, pp. 65-79.
Gavalas, A. 2002, "ArRAnging the hindbrain", Trends in neurosciences, vol. 25, no. 2, pp. 61-64.
Gilbert SF. Developmental Biology. 6th edition. Sunderland (MA): Sinauer Associates; 2000. Available from:
http://www.ncbi.nlm.nih.gov/books/NBK9983/
Gilbert SF. Developmental Biology. 9th edition. Sunderland (MA): Sinauer Associates; 2010
Giudicelli, F., Gilardi-Hebenstreit, P., Mechta-Grigoriou, F., Poquet, C. & Charnay, P. 2003, "Novel activities of Mafb
underlie its dual role in hindbrain segmentation and regional specification", Developmental biology, vol. 253, no. 1,
pp. 150-162.
Giudicelli, F., Taillebourg, E., Charnay, P. & Gilardi-Hebenstreit, P. 2001, "Krox-20 patterns the hindbrain through both
cell-autonomous and non cell-autonomous mechanisms", Genes & development, vol. 15, no. 5, pp. 567-580.
36
Glover, J.C., Renaud, J.S. & Rijli, F.M. 2006, "Retinoic acid and hindbrain patterning", Journal of neurobiology, vol. 66,
no. 7, pp. 705-725.
Gomaa, M.S., Lim, A.S.T., Wilson Lau, S.C., Watts, A., Illingworth, N.A., Bridgens, C.E., Veal, G.J., Redfern, C.P.F.,
Brancale, A., Armstrong, J.L. & Simons, C. 2012, "Synthesis and CYP26A1 inhibitory activity of novel methyl 3[4-(arylamino)phenyl]-3-(azole)-2,2-dimethylpropanoates", Bioorganic & medicinal chemistry, vol. 20, no. 20, pp.
6080-6088.
Gould, A., Itasaki, N. & Krumlauf, R. 1998, "Initiation of rhombomeric Hoxb4 expression requires induction by somites
and a retinoid pathway", Neuron, vol. 21, no. 1, pp. 39-51.
Hale, L.A., Tallafuss, A., Yan, Y.L., Dudley, L., Eisen, J.S. & Postlethwait, J.H. 2006, "Characterization of the retinoic
acid receptor genes raraa, rarab and rarg during zebrafish development", Gene expression patterns : GEP, vol. 6,
no. 5, pp. 546-555.
Hermsen, S.A., Pronk, T.E., van den Brandhof, E.J., van der Ven, L.T. & Piersma, A.H. 2012, "Concentration-response
analysis of differential gene expression in the zebrafish embryotoxicity test following flusilazole exposure",
Toxicological sciences : an official journal of the Society of Toxicology, vol. 127, no. 1, pp. 303-312.
Hernandez, R.E., Putzke, A.P., Myers, J.P., Margaretha, L. & Moens, C.B. 2007, "Cyp26 enzymes generate the retinoic
acid response pattern necessary for hindbrain development", Development (Cambridge, England), vol. 134, no. 1,
pp. 177-187.
Kane, M.A., Folias, A.E., Wang, C. & Napoli, J.L. 2010, "Ethanol elevates physiological all-trans-retinoic acid levels in
select loci through altering retinoid metabolism in multiple loci: a potential mechanism of ethanol toxicity", FASEB
journal : official publication of the Federation of American Societies for Experimental Biology, vol. 24, no. 3, pp.
823-832.
Kayam, G., Kohl, A., Magen, Z., Peretz, Y., Weisinger, K., Bar, A., Novikov, O., Brodski, C. & Sela-Donenfeld, D.
2013, "A novel role for Pax6 in the segmental organization of the hindbrain", Development, vol. 140, no. 10, pp.
2190-2202.
Kiecker, C. & Lumsden, A. 2005, "Compartments and their boundaries in vertebrate brain development", Nature Reviews
Neuroscience, vol. 6, no. 7, pp. 553-564.
Kim, F.A., Sing, l.A., Kaneko, T., Bieman, M., Stallwood, N., Sadl, V.S. & Cordes, S.P. 2005, "The vHNF1
homeodomain protein establishes early rhombomere identity by direct regulation of Kreisler expression",
Mechanisms of development, vol. 122, no. 12, pp. 1300-1309.
Kot-Leibovich, H. & Fainsod, A. 2009, "Ethanol induces embryonic malformations by competing for retinaldehyde
dehydrogenase activity during vertebrate gastrulation", Disease models & mechanisms, vol. 2, no. 5-6, pp. 295-305.
Krewski, D., Acosta Jr, D., Andersen, M., Anderson, H., Bailar III, J.C., Boekelheide, K., Brent, R., Charnley, G.,
Cheung, V.G. & Green Jr, S. 2007, "Summary" in Toxicity testing in the 21st century: a vision and a strategy
Taylor & Francis, pp. 1-17.
Kumar, A., Singh, C.K., DiPette, D.D. & Singh, U.S. 2010, "Ethanol impairs activation of retinoic acid receptors in
cerebellar granule cells in a rodent model of fetal alcohol spectrum disorders", Alcoholism, Clinical and
Experimental Research, vol. 34, no. 5, pp. 928-937.
Liu, J., Laufer, E. & Jessell, T.M. 2001, "Assigning the positional identity of spinal motor neurons: rostrocaudal
patterning of Hox-c expression by FGFs, Gdf11, and retinoids", Neuron, vol. 32, no. 6, pp. 997-1012.
Liu, A. & Joyner, A.L. 2001, "Early anterior/posterior patterning of the midbrain and cerebellum", Annual Review of
Neuroscience, vol. 24, pp. 869-896.
MacLean, G., Abu-Abed, S., Dolle, P., Tahayato, A., Chambon, P. & Petkovich, M. 2001, "Cloning of a novel retinoicacid metabolizing cytochrome P450, Cyp26B1, and comparative expression analysis with Cyp26A1 during early
murine development", Mechanisms of development, vol. 107, no. 1-2, pp. 195-201.
Maclean, G., Dolle, P. & Petkovich, M. 2009, "Genetic disruption of CYP26B1 severely affects development of neural
crest derived head structures, but does not compromise hindbrain patterning", Developmental dynamics : an official
publication of the American Association of Anatomists, vol. 238, no. 3, pp. 732-745.
Maden, M. 2006, "Retinoids and spinal cord development", Journal of neurobiology, vol. 66, no. 7, pp. 726-738.
37
Makki, N. & Capecchi, M.R. 2011, "Identification of novel Hoxa1 downstream targets regulating hindbrain, neural crest
and inner ear development", Developmental biology, vol. 357, no. 2, pp. 295-304.
Marrs, J.A., Clendenon, S.G., Ratcliffe, D.R., Fielding, S.M., Liu, Q. & Bosron, W.F. 2010, "Zebrafish fetal alcohol
syndrome model: effects of ethanol are rescued by retinoic acid supplement", Alcohol (Fayetteville, N.Y.), vol. 44,
no. 7-8, pp. 707-715.
Martinez-Barbera, J.P., Signore, M., Boyl, P.P., Puelles, E., Acampora, D., Gogoi, R., Schubert, F., Lumsden, A. &
Simeone, A. 2001, "Regionalisation of anterior neuroectoderm and its competence in responding to forebrain and
midbrain inducing activities depend on mutual antagonism between OTX2 and GBX2", Development (Cambridge,
England), vol. 128, no. 23, pp. 4789-4800.
McCaffery, P., Koul, O., Smith, D., Napoli, J.L., Chen, N. & Ullman, M.D. 2004, "Ethanol increases retinoic acid
production in cerebellar astrocytes and in cerebellum", Brain research.Developmental brain research, vol. 153, no.
2, pp. 233-241.
Menegola, E., Broccia, M.L., Di Renzo, F., Massa, V. & Giavini, E. 2005a, "Craniofacial and axial skeletal defects
induced by the fungicide triadimefon in the mouse", Birth defects research.Part B, Developmental and reproductive
toxicology, vol. 74, no. 2, pp. 185-195.
Menegola, E., Broccia, M.L., Di Renzo, F., Massa, V. & Giavini, E. 2005b, "Study on the common teratogenic pathway
elicited by the fungicides triazole-derivatives", Toxicology in vitro : an international journal published in
association with BIBRA, vol. 19, no. 6, pp. 737-748.
Menegola, E., Broccia, M.L., Di Renzo, F., Massa, V. & Giavini, E. 2004, "Relationship between hindbrain
segmentation, neural crest cell migration and branchial arch abnormalities in rat embryos exposed to fluconazole
and retinoic acid in vitro", Reproductive toxicology (Elmsford, N.Y.), vol. 18, no. 1, pp. 121-130.
Mic, F.A., Haselbeck, R.J., Cuenca, A.E. & Duester, G. 2002, "Novel retinoic acid generating activities in the neural tube
and heart identified by conditional rescue of Raldh2 null mutant mice", Development (Cambridge, England), vol.
129, no. 9, pp. 2271-2282.
Mollard, R., Viville, S., Ward, S.J., Decimo, D., Chambon, P. & Dolle, P. 2000, "Tissue-specific expression of retinoic
acid receptor isoform transcripts in the mouse embryo", Mechanisms of development, vol. 94, no. 1-2, pp. 223-232.
Niederreither, K., Vermot, J., Schuhbaur, B., Chambon, P. & Dollé, P. 2000, "Retinoic acid synthesis and hindbrain
patterning in the mouse embryo", Development, vol. 127, no. 1, pp. 75-85.
Niederreither, K., Subbarayan, V., Dolle, P. & Chambon, P. 1999, "Embryonic retinoic acid synthesis is essential for
early mouse post-implantation development", Nature genetics, vol. 21, no. 4, pp. 444-448.
Niederreither, K., Vermot, J., Fraulob, V., Chambon, P. & Dolle, P. 2002, "Retinaldehyde dehydrogenase 2 (RALDH2)independent patterns of retinoic acid synthesis in the mouse embryo", Proceedings of the National Academy of
Sciences of the United States of America, vol. 99, no. 25, pp. 16111-16116.
Olivera-Martinez, I. & Storey, K.G. 2007, "Wnt signals provide a timing mechanism for the FGF-retinoid differentiation
switch during vertebrate body axis extension", Development, vol. 134, no. 11, pp. 2125-2135.
Oosterveen, T., Niederreither, K., Dollé, P., Chambon, P., Meijlink, F. & Deschamps, J. 2003, "Retinoids regulate the
anterior expression boundaries of 5′ Hoxb genes in posterior hindbrain", The EMBO journal, vol. 22, no. 2, pp.
262-269.
Pasqualetti, M., Neun, R., Davenne, M. & Rijli, F.M. 2001, "Retinoic acid rescues inner ear defects in Hoxa1 deficient
mice", Nature genetics, vol. 29, no. 1, pp. 34-39.
Reijntjes, S., Gale, E. & Maden, M. 2004, "Generating gradients of retinoic acid in the chick embryo: Cyp26C1
expression and a comparative analysis of the Cyp26 enzymes", Developmental dynamics : an official publication of
the American Association of Anatomists, vol. 230, no. 3, pp. 509-517.
Ren, J., Xiong, X., Sha, Y., Yan, M., Lin, B., Wang, J., Jing, Y., Zhao, D. & Cheng, M. 2008, "Structure prediction and
R115866 binding study of human CYP26A1: homology modelling, fold recognition, molecular docking and MD
simulations", Molecular Simulation, vol. 34, no. 3, pp. 337-346.
Rhinn, M. & Dollé, P. 2012, "Retinoic acid signalling during development", Development, vol. 139, no. 5, pp. 843-858.
Rhinn, M. & Brand, M. 2001, "The midbrain--hindbrain boundary organizer", Current opinion in neurobiology, vol. 11,
no. 1, pp. 34-42.
38
Ribes, V., Le Roux, I., Rhinn, M., Schuhbaur, B. & Dolle, P. 2009, "Early mouse caudal development relies on crosstalk
between retinoic acid, Shh and Fgf signalling pathways", Development (Cambridge, England), vol. 136, no. 4, pp.
665-676.
Robinson, J.F., Tonk, E.C., Verhoef, A. & Piersma, A.H. 2012, "Triazole induced concentration-related gene signatures
in rat whole embryo culture", Reproductive toxicology (Elmsford, N.Y.), vol. 34, no. 2, pp. 275-283.
Sakai, Y., Meno, C., Fujii, H., Nishino, J., Shiratori, H., Saijoh, Y., Rossant, J. & Hamada, H. 2001, "The retinoic acidinactivating enzyme CYP26 is essential for establishing an uneven distribution of retinoic acid along the anterioposterior axis within the mouse embryo", Genes & development, vol. 15, no. 2, pp. 213-225.
Santos-Ledo, A., Cavodeassi, F., Carreño, H., Aijón, J. & Arévalo, R. 2013, "Ethanol alters gene expression and cell
organisation during optic vesicle evagination", Neuroscience, .
Serpente, P., Tumpel, S., Ghyselinck, N.B., Niederreither, K., Wiedemann, L.M., Dolle, P., Chambon, P., Krumlauf, R.
& Gould, A.P. 2005, "Direct crossregulation between retinoic acid receptor {beta} and Hox genes during hindbrain
segmentation", Development (Cambridge, England), vol. 132, no. 3, pp. 503-513.
Sirbu, I.O., Gresh, L., Barra, J. & Duester, G. 2005, "Shifting boundaries of retinoic acid activity control hindbrain
segmental gene expression", Development (Cambridge, England), vol. 132, no. 11, pp. 2611-2622.
Sunmonu, N.A., Li, K., Guo, Q. & Li, J.Y. 2011, "Gbx2 and Fgf8 are sequentially required for formation of the
midbrain-hindbrain compartment boundary", Development (Cambridge, England), vol. 138, no. 4, pp. 725-734.
Tahayato, A., Dolle, P. & Petkovich, M. 2003, "Cyp26C1 encodes a novel retinoic acid-metabolizing enzyme expressed
in the hindbrain, inner ear, first branchial arch and tooth buds during murine development", Gene expression
patterns : GEP, vol. 3, no. 4, pp. 449-454.
Thatcher, J.E., Buttrick, B., Shaffer, S.A., Shimshoni, J.A., Goodlett, D.R., Nelson, W.L. & Isoherranen, N. 2011,
"Substrate specificity and ligand interactions of CYP26A1, the human liver retinoic acid hydroxylase", Molecular
pharmacology, vol. 80, no. 2, pp. 228-239.
Theunissen, P.T., Pennings, J.L., van Dartel, D.A., Robinson, J.F., Kleinjans, J.C. & Piersma, A.H. 2013,
"Complementary Detection of Embryotoxic Properties of Substances in the Neural and Cardiac Embryonic Stem
Cell Tests", Toxicological Sciences, vol. 132, no. 1, pp. 118-130.
Theunissen, P.T., Robinson, J.F., Pennings, J.L., de Jong, E., Claessen, S.M., Kleinjans, J.C. & Piersma, A.H. 2012,
"Transcriptomic concentration-response evaluation of valproic acid, cyproconazole, and hexaconazole in the neural
embryonic stem cell test (ESTn)", Toxicological sciences : an official journal of the Society of Toxicology, vol. 125,
no. 2, pp. 430-438.
Uehara, M., Yashiro, K., Mamiya, S., Nishino, J., Chambon, P., Dolle, P. & Sakai, Y. 2007, "CYP26A1 and CYP26C1
cooperatively regulate anterior-posterior patterning of the developing brain and the production of migratory cranial
neural crest cells in the mouse", Developmental biology, vol. 302, no. 2, pp. 399-411.
Vendrell, V., Vazquez-Echeverria, C., Lopez-Hernandez, I., Alonso, B.D., Martinez, S., Pujades, C. & Schimmang, T.
2013, "Roles of Wnt8a during formation and patterning of the mouse inner ear", Mechanisms of development, vol.
130, no. 2-3, pp. 160-168.
Wallingford, J.B., Niswander, L.A., Shaw, G.M. & Finnell, R.H. 2013, "The Continuing Challenge of Understanding,
Preventing, and Treating Neural Tube Defects", Science, vol. 339, no. 6123.
Wassef, M.A., Chomette, D., Pouilhe, M., Stedman, A., Havis, E., Desmarquet-Trin Dinh, C., Schneider-Maunoury, S.,
Gilardi-Hebenstreit, P., Charnay, P. & Ghislain, J. 2008, "Rostral hindbrain patterning involves the direct activation
of a Krox20 transcriptional enhancer by Hox/Pbx and Meis factors", Development (Cambridge, England), vol. 135,
no. 20, pp. 3369-3378.
White, J.C., Highland, M., Kaiser, M. & Clagett-Dame, M. 2000, "Vitamin A deficiency results in the dose-dependent
acquisition of anterior character and shortening of the caudal hindbrain of the rat embryo", Developmental biology,
vol. 220, no. 2, pp. 263-284.
White, J.C., Shankar, V.N., Highland, M., Epstein, M.L., DeLuca, H.F. & Clagett-Dame, M. 1998, "Defects in
embryonic hindbrain development and fetal resorption resulting from vitamin A deficiency in the rat are prevented
by feeding pharmacological levels of all-trans-retinoic acid", Proceedings of the National Academy of Sciences, vol.
95, no. 23, pp. 13459-13464.
39
Wilson, V., Olivera-Martinez, I. & Storey, K.G. 2009, "Stem cells, signals and vertebrate body axis extension",
Development, vol. 136, no. 10, pp. 1591-1604.
Wolpert, L., Jessell, T., Lawrence, P., Meyerowitz, E., Robertson, E. & Smith, J. 2007, Principles of development, 3rd
edn, Oxford University Press Oxford.
Wong, E.Y., Wang, X.A., Mak, S.S., Sae-Pang, J.J., Ling, K.W., Fritzsch, B. & Sham, M.H. 2011, "Hoxb3 negatively
regulates Hoxb1 expression in mouse hindbrain patterning", Developmental biology, vol. 352, no. 2, pp. 382-392.
Ybot-Gonzalez, P., Cogram, P., Gerrelli, D. & Copp, A.J. 2002, "Sonic hedgehog and the molecular regulation of mouse
neural tube closure", Development, vol. 129, no. 10, pp. 2507-2517.
Yelin, R., Kot, H., Yelin, D. & Fainsod, A. 2007, "Early molecular effects of ethanol during vertebrate embryogenesis",
Differentiation; research in biological diversity, vol. 75, no. 5, pp. 393-403.
Yelin, R., Schyr, R.B., Kot, H., Zins, S., Frumkin, A., Pillemer, G. & Fainsod, A. 2005, "Ethanol exposure affects gene
expression in the embryonic organizer and reduces retinoic acid levels", Developmental biology, vol. 279, no. 1, pp.
193-204.
Young, T. & Deschamps, J. 2009, "Hox, Cdx, and Anteroposterior Patterning in the Mouse Embryo", Current topics in
developmental biology, vol. 88, pp. 235-255.
40