Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
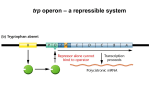
Regulatory Signatures of Cancer Cell Lines Inferred from Expression Data 1,2 Krishnan , 2 Ma’ayan Jayanth (Jay) Avi 1Mahopac High School, Mahopac, NY 10541 2Systems Biology Center New York and Department of Pharmacology and Systems Therapeutics, Mount Sinai School of Medicine, New York NY 2 Abstract While gene expression data is widely available describing mRNA levels in different cancer cells lines, the molecular regulatory mechanisms responsible for these changes are still poorly understood. Here we developed a rationale approach to infer regulatory mechanisms governing changes in gene expression by integrating datasets of protein/DNA interactions, proteinprotein interactions and kinase-substrate interactions collected from prior biological knowledge. We first utilize data obtained from genome-wide ChIP-on-chip and ChIP-Seq experiments to connect mRNA expression levels of the NCI-60 cancer cell lines to the transcription factors most likely regulating them. These identified transcription factors are then “connected”, using known protein-protein interactions, to form cancer specific sub-networks. Within these sub-networks we assess the enrichment for protein kinase substrates to infer the protein kinases likely regulating these complexes. Finally, using quantitative comparison of the up and down regulated genes for each cancer cell line, and genes affected by FDA approved drugs applied to cancer cells, we predict the mechanisms of action of these drugs. Following this path, from changes in gene expression to transcription factors to protein kinases we can provide a more thorough understanding of the regulatory mechanisms behind the observed mRNA levels in the NCI-60 cancer cell lines and other cancer cells. This approach proposes mechanisms of action for drugs. Wet lab experimental validation of this approach is still necessary, it can be done using single drugs or combinations of them. Introduction • The NCI-60 database provides mRNA profiles from microarray experiments of 60 commonly studies cancer cell lines • Although analyzing these mRNA values is a reliable method to measure the mRNA level of many genes within a cell, this method offers little clues about how cells are regulated • While mRNA profiles indicates changes caused by cancer, understanding the underlying regulatory mechanisms disregulated in different cancers will bring us closer to therapeutics • In this project we aim to identify the transcription factors, protein complexes and protein kinases responsible for the aberrant expression of genes in the various types of cancer cell lines Workflow Top ranked transcription factors most likely responsible for the observed changes in expression Microarray Analyze mRNA profile from NCI -60 database by using statistical techniques to compute over/under expressed genes Example of Process Identify protein sub-networks that “connect” the transcription factors through additional proteins Wet lab experimental validation Top ranked protein kinases most likely regulating the protein sub-networks PLD1 SART1 WARS DNAJB1 M6PR RAE1 GPAA1 SLC29A1 IL13RA1 TRAK2 CHPF GAS7 CUL4B SLC6A8 CSNK1E PRKCD DHPS SLC37A4 TCTA TIMP2 CTSF CD302 KAT2B EDNRB NOV GJB1 AP1S1 ATP7A EIF2S3 DCT NPAS2 SCRG1 TRAIP UBFD1 TYRP1 HBE1 RXRG PLXNB3 KCNS3 S100A3 TYR GLRX HCG4 GPR143 DGKI STAU1 MAGEA1 CTAG2 ZNF200 CAST HLA-DRA TXN2 TNFRSF14 FAM3A RAB27A PLP1 HSP90AA1 MAGEA12 CTAG1B NRP2 SPIN2B C1orf144 SLC25A6 ZCCHC24 SUCLG2 C14orf109 CLCN2 C9orf61 CCPG1 MAGEA2B DPY19L2P2 DUSP10 SLC6A10P SLC5A4 GK3P BACE2 MUL1 METT11D1 MRPS18A KLF11 CRIPT MTO1 HEY1 FBXL15 CSGALNACT1 CADPS2 PRR7 GAL3ST4 NUDT11 CEP97 MGAT4B FAM86C ROPN1B C20orf30 TRIM48 RPL23AP7 MICALL1 LDLRAP1 C17orf90 LUZP1 LOC348926 PCOTH FAM86B1 LPCAT2 SURF4 HAGHL TNFSF13B FAM167B SPRYD5 DGAT2 ULK3 TOMM40L FAM160B1 SNX30 TMEM55A HDAC10 LOC400657 AFAP1L1 FAM125A OLIG1 HSD11B1L SCARNA15 SMYD4 LOC153364 CAMK2N2 CHRM1 AARS2 ANKRD54 KIAA1524 KIAA1586 GPR158 SLITRK4 GNASAS ELOVL3 ST6GALNAC3 RNF175 C5orf35 LOC147645 LOC730259 TMEM171 GPNMB SREBF2 SEC11A AKR7A2 USF2 CNOT8 GTF2H1 PCOLCE CHMP2B STX7 SNTA1 PTPN18 HPS5 SMCR7L ACP5 DYNC1I1 BCHE GSTT2 TRPM2 DDX18 SLC4A3 ASPA SLC22A18AS PPP2R4 CGGBP1 CSRP2 SLC25A11 PDIA6 BEST1 MCM7 SLC1A4 KHDRBS3 ART3 CAPN3 GYG2 DLAT TUBB4 RFNG MORC3 MAGEA5 AZI1 UAP1L1 SFXN3 HLA-DMA ALDH18A1 ARHGEF3 FAHD2A RINT1 ITIH5 CA14 C14orf139 LONRF3 TINF2 TP53TG3 TH1L C5orf54 CDCA3 C3orf64 GPR177 COL9A1 XPO5 PDXP COPG2 C2orf30 C6orf89 UBL7 GGT7 C12orf34 C3orf38 C11orf82 ENHO CITED4 PAGE2 LOC730124 GBGT1 ZC3H12C FSTL5 CLEC2L LRRC33 NPHP3 HMCN1 DLX1 Top 222 over expressed genes for cancer cell line MDA_N (melanoma) Future Research Identify protein sub-networks that “connect” the transcription factors through additional proteins Analyzing the mRNA profile from the NCI-60 database Statistical Methods: Probes 1 2 … n 1 M1,1 M2, 1 … Mn,1 2 M1,2 M2,2 … Mn,2 Cancer cell lines 3 … c M1,3 … M1,c M2,3 … M2,c … … … Mn,3 … Mn,c With gene input, ChEA identified the top ranked transcription factors Genes2Networks output of protein subnetworks when top 10 transcription factors from ChEA were given as an input … … … … … Genes2Networks KEA ChEA, Genes2Networks, and KEA are all web-based tools developed at the Ma’ayan lab to allow users to predict which transcription factors, protein subnetworks, and protein kinases are most correlated with their inputted seed list • By using the identified up and down regulated genes for each cancer cell line as an input for ChEA; the top ranked transcription factors (based on p-value from Fisher’s Exact Test) that most likely influence the input seed list are given as the output Genes2Networks •The transcription factor output for each cancer cell line from ChEA is used as an input to Genes2Networks • Genes2Networks connects lists of transcription factors with other protein intermediates from mammalian protein interactions databases KEA Wet lab experimental validation • Differentially expressed gene lists from the various NCI-60 Cancer Cell Lines are used as input. • Over expressed and under expressed genes are identified for specific cancer cell lines • The following algorithm was implemented: • The NCI-60 database was parsed and 18,133 unique genes were identified • The population mean for the expression of each of the genes across all the 60 cancer cell lines was calculated • The sample mean and sigma for each (gene, cancer cell line) pair was calculated • The two-sided T-test statistic was applied for each (gene, cancer cell line) pair. • Whether the gene was over expressed or under expressed was calculated by checking whether the test statistic exceeded a critical T score or was a less than a critical T score determined based on a particular P value. • A list of genes which are over/under expressed for multiple cancer cell lines was developed ChEA 60 M1,60 M2,60 … Mn,60 Population mean µ = ∑ Mi,j / (n * 60) i=1,n; j=1,60 Sample mean of gene expressions for cancer cell line “c” = x̄ = ∑ Mi,c / n i=1,n •The unique protein sub-networks outputted by Genes2Networks can then be inputted into KEA which identifies protein kinases most likely regulating the proteins from the subnetwork using the Fisher’s Exact Test. • At this stage top regulating transcription factors, protein sub-networks and kinases have been identified for each of the NCI-60 cancer cell lines • An integrated matrix can now be created in order to holistically compare the data by displaying the top regulating elements and their putative effects on the different cell lines Future Research • Future research involves further analyzing other cancer datasets • Cluster analysis will be done to groups transcription factors or kinases that were identified • Additionally, by combining such data with data collected for drug perturbation of these cells, we may be able to suggest which drugs can reverse the observed changes Acknowledgements Std deviation of gene expressions for cancer cell line “c” = s = sqrt (∑ (Xi – x̄ ) 2 / (n-1)) i=1,n Test statistic = ( xbar – µ) * sqrt(n) / s Top ranked kinase proteins identified from KEA This research was supported by NIH Grant No. 5P50GM071558