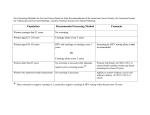
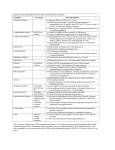
Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Genetic Screening: Carriers and Affected Individuals Linda L. McCabe1,2 and Edward R.B. McCabe1,2,3 1 Departments of Human Genetics and Pediatrics, David Geffen School of Medicine at UCLA 2 UCLA Center for Society, the Individual and Genetics 3 Mattel Children’s Hospital at UCLA Linda L. McCabe, Ph.D. Department of Human Genetics David Geffen School of Medicine at UCLA 695 Charles Young Drive South, 4506 Gonda Los Angeles, California 90095 [email protected] Edward R.B. McCabe, M.D., Ph.D. Department of Pediatrics David Geffen School of Medicine at UCLA 10833 Le Conte Ave., 22-412 MDCC Los Angeles, California 90095-1752 [email protected] Key words: genetic screening; technology; newborn screening; carrier; affected Proofs should be sent to: Edward R.B. McCabe, M.D., Ph.D. Department of Pediatrics David Geffen School of Medicine at UCLA 10833 Le Conte Ave., 22-412 MDCC Los Angeles, California 90095-1752 Telephone: 310-825-5095 Fax: 310-206-4584 [email protected] ABSTRACT Genetic screening utilizes analytical approaches adapted for high throughput to identify carrier and affected individuals in a targeted population. Genetic screening is focused currently on carrier screening, prenatal screening, and newborn screening. Newborn screening should serve as a model for all genetic screening, with more than forty years of experience and numerous lessons learned. Just as in all of genetic screening, there are policy concerns in newborn screening regarding which disorders and technologies should be selected, and how centralized or decentralized should be the process to set policy. The need to share experiences and develop databases transcends all of genetic screening. The future will see population-based screening for adult onset disorders. However, there will need to be extensive research to define predictive risk for various ethno-cultural groups and to determine effective interventions. Ethical concerns regarding the timing of population screening will need to be resolved if genomic medicine will achieve its promise of a predictive, preventive, and personalized medicine. Genetic screening involves identification of individuals affected with a disease, at risk for developing disease, or at risk for having a child affected with a disease. It involves a population-based approach, and populations are selected with specific goals and strategies in mind. Because testing of entire populations is involved, the analytical methodologies used in genetic screening are adapted for high throughput, maximum efficiency, and lowest cost. Decisions regarding test selection must be consistent with optimized ascertainment of true positives, while keeping false negatives to absolute lowest possible frequency, and minimizing false positives (41, 50) Currently genetic screening in the United States involves carrier screening within selected populations, prenatal screening of women who receive prenatal care and are willing to participate, and newborn screening for nearly all newborns. In the future, genetic screening will move the practice of medicine from acute responses at the time of a medical emergency, to a public health, predictive strategy. In the era of predictive medicine, entire populations, or targeted groups, will be screened for common disorders, (e.g. cancer, diabetes, heart disease), using genetic tests. The goal will be to prevent these disorders in those at risk, through the use of diet, exercise, or medication, and to ensure increased monitoring of those at risk (25). If genetic screening for disease is successful, then it will identify individuals with a disease, or at risk for disease, before the onset of symptoms, and at a time that interventions will be effective. Screening intended to identify unaffected carriers has the goal of providing information to individuals so that they will understand their reproductive options. Carrier screening, however, may be a by-product of disease screening, e.g., heterozygotes in the course of hemoglobinopathy. In that case, the carriers may be ascertained without specifically requesting this information, and at a time and/or under circumstances when knowledge of, interest in, or access to genetic counseling is low. Newborn screening serves as a model for all of genetic screening (25, 37). Newborn screening identifies affected individuals and carriers. Since an individual’s carrier status will not be useful to them until they reach reproductive age, concern has been raised about the maintenance and fidelity of the carrier information, so that it will be available and accurate for reproductive decision-making. History of Newborn Screening Newborn screening was developed as a “low tech” enterprise, and its origins demonstrate that a single individual, Robert Guthrie, M.D., Ph.D., can make a difference (26). Guthrie was an investigator who utilized a bacterial inhibition assay for the measurement of amino acid concentrations. This methodology, the bacterial inhibition assay (BIA), involved measuring the growth zone of bacteria around a paper disc, containing the amino acid of interest. The size of the growth zone was proportional to the concentration of the analyte, and the BIA was “turned” to the concentrations anticipated in the sample by use of an inhibitor that competed with the natural analyte and could not be used by the bacteria to support growth and cell division. He showed that phenylalanine in a dried blood spot could be measured by the BIA (21). Motivated by a son with mental retardation and a niece with phenylketonuria (PKU), Guthrie organized parents in state Associations for Retarded Citizens (ARCs), in the 1960s, to lobby their state legislators to establish newborn screening for PKU. The goal was to prevent mental retardation in children with PKU by initiation of a special diet that was restricted in phenylalanine. In addition to developing the BIA to screen for hyperphenylaninemia and other amino acidopathies, Guthrie also recognized the value of dried blood specimens, now known as “Guthrie cards”, for transport of samples from the birthing hospital to a centralized newborn screening laboratory. When testing for a rare disorder like PKU, (estimated frequency at that time of 1/10,000 – 1/12,000 live births), the average hospital would only identify an affected patient once over several years or longer. A centralized, dedicated laboratory would have more experience, better quality assurance, and this would decrease the risk of false negatives. Newborn screening dried blood specimens have a number of additional advantages. Once the blood is dry, the analytes (small molecules, proteins and DNA) are stable. They are easily transported and stored. Most pathogens are killed when dry. The drawbacks to this form of sample are the specialized equipment required for sample handling and the systems required for storage and retrieval. However, dried blood samples will continue to be used since newborn screening systems find them so easy to work with. Because DNA and protein are stable in these specimens, the states’ collection of newborn screening blood blotters represent a biobank. The fact that few states have laws governing the use and disposition of newborn screening samples is concerning (29). With the addition of new disorders came new newborn screening technologies. Technologies currently used for newborn screening, in addition to the Guthrie test (BIA), include: enzyme assays; immunoassays (RIAs and EIAs); electrophoresis; high performance liquid chromatography (HPLC); tandem mass spectrometry (MS/MS); and DNA confirmatory testing. The addition of the newer methodologies shows that the formerly low-tech enterprise of newborn screening is moving into the high tech world. Tandem Mass Spectrometry (MS/MS) The addition of MS/MS to newborn screening has greatly increased the number of disorders tested (8, 43). Mass spectrometry involves ionization and adsorption of excess energy by individual molecules, resulting in fragmentation (dissociation), and then mass analysis. MS/MS provides serial fragmentation that with computerized analysis permits identification of signature metabolites for individual inborn errors of metabolism. MS/MS is used to screen for amino acid disorders including PKU, hypertyrosinemia, Maple Syrup Urine Disease (MSUD), homocystinemia, hyperargininemia, citrullinemia, and argininosuccinic acidemia. Organic acid disorders such as methylmalonic acidemia, propionic acidemia, isovaleric acidemia, and the glutaric acidemias are also detected. MS/MS can also screen for fatty acid oxidation disorders like medium chain acyl CoA dehydrogenase deficiency (MCAD). MS/MS has the potential for expanded applications in screening for steroids, such as cholesterol and steroid precursors, hormones, purines and pyrimidines, fatty acids, and toxins. Wilcken et al. (56) compared rate of detection of amino acid, organic acid, and fatty acid oxidation disorders, before (1974-1998) and after (1998-2002) the introduction of MS/MS to the newborn screening program in New South Wales, Australia. MS/MS increased the rate of detection for MCAD and other fatty acid oxidation disorders. The costs of tandem mass spectrometry were $.70/newborn, $217/ confirmed diagnosis, and $2,519/ case detected. The experience with MS/MS in Germany was described by Schulze et al. (49). They screened 250,000 newborns for 23 disorders using MS/MS. The false positive rate was 0.33%. They found 106 true positives after confirmatory tests. MS/MS screening benefited 61 of the 106 true positives and increased the detection rate by two-fold. Chace et al. (8) investigated 793 deaths of children under the age of three, in Virginia, in a study carried out in collaboration with the state’s medical examiners. Eight, or one percent, had an inborn error of metabolism diagnosed by MS/MS, using dried blood specimens obtained postmortem. Four had fatty acid oxidation disorders (two were MCAD) and four had organic acid disorders. While one of these eight children died on the second day of life, the authors conducted that the other seven would have benefited from newborn screening with MS/MS. In a similar study, Wilcox and et al. (57) tested newborn blood spots from Oregon infants who experienced sudden unexpected death for fatty acid oxidation disorders, using MS/MS. One percent of the infants had fatty acid oxidation disorders: two had MCAD and one had VLCAD. Schoen et al. (48) performed a cost-benefit analysis of MS/MS newborn screening at Kaiser in Northern California. The cost was $5,827 per quality-adjusted life year saved by MS/MS screening. The authors concluded that this compared favorably with screening for adult disorders such as breast cancer and prostate cancer. Insinga et al. (23) analyzed cost effectiveness of MS/MS in the Wisconsin newborn screening program. They found a cost of $6,008 per quality adjusted life year saved by testing for 14 fatty acid oxidation and organic acid disorders. This technology remained cost-effective if the cost to add the tests was less than $13.05 per test. From this research we conclude that ascertainment of infants with inherited metabolic disorders increases with MS/MS. Without MS/MS these affected infants would die, many without an antemortem diagnosis. MS/MS is cost-effective. Newborn screening is more than the test, but rather a system including, for example: in the preanalytic phase, education, sample procurement, and transport; in the analytic phase, accessioning, testing, and result posting; and in post-analytic phase, result distribution, diagnostic confirmation, therapeutic intervention, genetic counseling, long-term followup, and programmatic quality assurance (44). Organized, protocol-driven, long-term follow-up of affected infants is required (32). DNA Confirmatory Testing McCabe et al (34) found that DNA could be extracted from the dried blood spot on a newborn screening blotter for Southern blot analysis. This led to the military “DNA dog tag” for identification and specimen collection for criminal forensics. It also made possible 2nd tier DNA follow-up and confirmation from the original newborn screening specimen and the possibility of primary DNA screening for specific mutations. Applying the polymerase chain reaction (PCR) to DNA from newborn screening blood blotters, Jinks et al. (24) demonstrated the feasibility of molecular genetic diagnosis of sickle cell disease. They argued that this methodology would decrease follow-up costs, because the original blotter could be used, and that more timely information could be provided because the persistence of fetal hemoglobin that complicated the electrophoretic analysis was not an issue for DNA diagnosis. Rubin et al. (47) showed that the DNA in the newborn screening sample was stable for up to one year, facilitating DNA analysis for sickle cell disease. Williams et al. (58), showed DNA stability in a newborn screening specimen for more than 15 years. Methods were improved to decrease time and cost of DNA follow-up (33). Descartes (13) demonstrated the feasibility of using DNA follow-up as part of a newborn screening program. Zhang and McCabe (60) showed that RNA could be extracted from the dried blood blotter, enabling diagnosis of other hemoglobinopathies, such as the β-thalassemias. Zhang et al. (61) showed that DNA follow-up halved the age at confirmed diagnosis, reducing the age from four months to two months. At the time, it was recommended that infants with sickle cell disease begin penicillin prophylaxis by four months of age to prevent the morbidity and mortality associated with infection (14). The cost of the DNA follow-up testing for hemoglobinopathies was estimated to be $10 – 25 per newborn (61), though another group carrying out second tier DNA testing for CF estimated their costs to be lower, at $2-3 (17). Use of DNA follow-up provides information regarding carrier status in addition to identifying the affected infants. If an infant is a carrier, one or both parents must be carriers. This information is immediately useful to the parents if they did not know about their own carrier status. Preserving this information regarding a newborn and providing it at the appropriate time for reproductive decision-making can be a concern. DNA testing has been used for newborn screening confirmation for a number of disorders including the hemoglobinopathies, cystic fibrosis, MCAD deficiency, and congenital adrenal hypoplasia (37). A variety of molecular genetic technologies are being used for neonatal hemoglobinopathy screening (5). These include restriction enzyme analysis, dot blot with allele specific oligonucleotide (ASO) hybridization, reverse dot blot, amplification refractory mutation system (ARMS), gap-PCR, SSCP and heteroduplex analysis, denaturing gradient gel electrophoresis (DGGE), automated sequencing, real-time PCR, and oligonucleotide microarray analysis. Need for Standardization of the Tests and Technologies of Newborn Screening According to the Government Accounting Office (16) all states screening for PKU and congenital hypothyroidism. Beyond these two tests, there is not programmatic uniformity. States test for four to 36 disorders. Most states screen for eight or fewer disorders. The Newborn Screening Task Force Report (44) called for a national agenda for state newborn screening programs regarding the disorders screened and the technologies utilized. The report emphasized that newborn screening is a system of care, not simply the analytical phase. There is a need to modernize the methodologies for newborn screening. There should be one platform, or at least a limited number of platforms, for analytes. The technology should be transparent to the user and the cost should be low. Nanotechnology will provide possible platforms, with microfluidic sample preparation and chip-based analyses, including amperometric measurements of DNA, proteins, and metabolites (15), and MS, MS/MS and MALDI-TOF on a chip (55). In addition to standardization of technologies, new screening programs should borrow from the successful newborn screening programs. When newborn hearing screening was developed during the 1990s, it developed independently in many hospitals and states, from established newborn screening programs (38). Newborn hearing screening is hospital-based, while newborn screening programs are centralized through the state newborn screening program. Hospitals do not have experience with follow-up for an initial positive hearing screen. There is a lack of audiologists trained in newborn hearing testing and diagnosis. Association of newborn hearing screening with the longstanding state newborn screening programs would provide needed infrastructure for diagnostic follow-up testing. Software programs, including digital data communications protocols, exist that would permit centralized quality assurance of hospital-based hearing screening. In addition, linkage of hearing screening to state heelstick screening programs would also enable a link between the hearing test and the newborn screening blood blotter that would facilitate DNA testing for a major cause of hearing loss, connexin-26 mutations. Mutations in the connexin-26 gene occur in 3% of the population and are responsible for 40% of all cases of childhood hearing loss (9). A single mutation is responsible for most of these cases in a mixed U.S. population (9), though, for example, among Ashkenazi Jews, a different mutation predominates (40). Additional mutations need to be assessed in either population in order to assure access to this technology by all populations (10, 11, 30). Hearing screening followed by DNA testing of the newborn screening blotter could prove to be more rapid and cost-effective than hearing screening followed by diagnostic audiometry, but requires linkage of these programs that in many states operate completely independently. Newborn Screening is the Model for Genomic Medicine Genomic medicine promises a medical paradigm that will be proactive, rather than reactive, and will provide care that is predictive, preventive, and personalized. Newborn screening has been guided by these principles from its beginning and is the laboratory for genomic medicine. We must move newborn screening into the modern era technologically, with expansion of MS/MS and other MS-related capabilities, and incorporation of nanotechnology solutions to sample preparation and analyses. To extend genetic screening beyond the newborn period will require careful development of programs, in addition to technologies. Mitchell et al. (39) described a long-standing program in Montreal that provided targeted screening of carrier status for Tay-Sachs disease and beta-thalassemias among high school students. As part of their biology coursework, students learned about these two disorders. They were offered carrier testing, and retained and used the information in their reproductive decisionmaking. High school, like the newborn period provides an opportunity to offer screening programs to a broad segment, if not 100% of the population. In the United States we would need to change our approach to age at independence for health care decisionmaking in order for screening to be effective in high schools (36). We would also need to reinforce legislative protections against the possibility of genetic discrimination. The American Academy of Pediatrics (AAP) Committee on Bioethics (1) argued against carrier testing in individuals under 18 years of age, unless pregnant or planning a pregnancy. They also opposed to predictive screening for adult-onset disorders before 18 years of age. The AAP Committee on Genetics (3) stated that those under 18 years of age should have molecular genetic testing if they would receive immediate medical benefits, or if another family member would benefit and there would be no harm to the person being tested. This AAP committee also stressed the importance of genetic counseling before and after the test. Population-Based Screening Beyond the Newborn Period Hereditary hemochromatosis can be improved with phlebotomy (59). However, a Center for Disease Control-National Human Genome Research Institute Expert Panel (6) concluded that DNA testing should not be used to screen for hereditary hemochromatosis. The Panel was concerned that there was a need to obtain more data on prevalence and penetrance of the mutations, and to evaluate the benefits of treatment for those with mutations. The Panel raised the possibility of genetic discrimination against those with mutations. A recent study (4) showed a very low penetrance. Only one of 152 individuals, who was homozygous for the mutation with the highest risk of hereditary hemochromatosis, showed any symptoms. A mutation that leads to Factor V Leiden, results in thrombophilia, i.e., an increased tendency to form blood clots. Vandenbroucke et al. (52) found that the Factor V Leiden mutation led to a seven-fold increase in the risk of venous thrombosis. They observed that the use of oral contraceptives led to a four-fold increased risk; however, women with the mutation, taking oral contraceptives, had their risk increased 30-fold. Since the absolute risk of venous thrombosis is low (28/10,000 person years), Vandenbroucke et al. (53) concluded that 500,000 women would need to be screened for Factor V Leiden, and tens of thousands would be denied oral contraceptives to prevent a single death. The American College of Medical Genetics found that there was no consensus on screening women before prescribing oral contraceptives (20). They did not recommend Factor V Leiden screening for healthy women planning to use oral contraceptives. A National Institutes of Health Consensus Development Conference on cystic fibrosis screening (42) recommended that the following individuals undergo mutation screening for cystic fibrosis: adult family members of patients with cystic fibrosis; partners of patients with cystic fibrosis; couples planning a pregnancy; and couples seeking prenatal care. With more than 900 mutations reported (19), a decision needed to be made whether to screen for all identified mutations or to screen for the most prevalent mutations. In either case, some cystic fibrosis carriers would be missed. An American College of Medical Genetics/American College of Obstetricians and Gynecologists/National Institutes of Health committee made recommendations for carrier screening for cystic fibrosis (18). The committee recommended screening for 25 mutations, selecting the recommended mutations as those that had a frequency of 0.1%. Members of ethnocultural groups should be told of the limitations of this test to detect carriers in their groups (e.g., African-Americans and Latinos) or of the low frequency of cystic fibrosis in their group (e.g., Asians and Native Americans). Mutations in the cystic fibrosis gene may be associated with phenotypes distinct from the classical pulmonary and nutritional manifestations, including azoospermia in men (31) and chronic sinusitis (45, 54). Prevention is one goal of screening for adult onset disorders such as Type 2 diabetes mellitus (46). One form of Type 2 diabetes is maturity onset diabetes of the young (MODY) (27). A diagnosis of MODY relies on three generations with autosomal dominant diabetes and two patients with onset 25 years of age. Lehto et al. (28) screened family members of Scandinavian patients with MODY for four genes. Thirteen percent had a mutation in one of these genes. If this screening could be performed before adolescence, there would be the opportunity to prevent the obesity associated with MODY through diet and exercise. Many of the interventions for disorders, like diabetes mellitus, obesity, heart disease, and even some forms of cancer, will involve lifestyle changes (32). These will included exercise and diet, and will involve changes in patterns of behavior throughout the individual’s life. This will represent a significant therapeutic challenge to successful screening programs for adult-onset disorders. Just as newborn screening is a system in which the screening test will be only one small part, so too will screening programs for later onset disorders be systems, and not simply the tests. Summary and Conclusions Informed population screening applies technology for the benefit of populations and the individuals composing those populations, and avoids health care disparities (37). There must be adequate information for each ethno-cultural group regarding mutation frequency and penetrance. In the absence of such information, there cannot be effective genetic counseling concerning the clinical utility of the test results. To avoid health care disparities, screening should be offered at a reasonable price or as part of a free public health program (51). The public needs to be educated regarding the risks and benefits of population screening (37). An understanding of the difference between carrier status and diagnosis as an affected individual must be a goal in preparing the public adequately for screening. Members of the public and health care professionals must be offered the opportunity to be educated regarding the predictive nature of genetic information, since that is so different from the usual absolute positive or negative result from a medical test. They should understand that the storage of their screening samples represents a DNA database (37). Since information in genetics and genomics is continuing to undergo development, participants in screening programs should be assured that they will receive information in the future, regarding unexpected disease associations related to the condition for which they are screened. The public should understand that a guiding principle of screening is that the participants benefit from presymptomatic testing. Members of the public should be actively involved in establishing screening policy (44). Health care providers also need education regarding genetic screening. They need to understand that screening programs will miss carriers and affected individuals (2). A negative screening result should never outweigh clinical acumen. If an individual has symptoms of a disorder, they should receive a diagnostic test. Screening test results are not infallible due to biologic, clerical and laboratory errors (12, 22, 35). Successful genetic screening to identify carriers and affected individuals will require reliance on principles derived from the most frequent genetic testing to date – the screening of the 4 million newborns each year in the U.S. and even more around the world. Extensive research will be required to determine the penetrance of mutations and the relative risks to different ethnocultural groups. The age of participants and the venue of the screening will have to be considered to ensure that screening is cost-effective and available to as many members of the target group as possible. Finally, research to determine effective prevention strategies, treatments, and/or effective focused screening for those at risk will be required to justify genetic screening. Disclosure: One of the authors (ERB McCabe) is an advisor to the nanotechnology company, GeneFluidics. Literature Cited 1. American Academy of Pediatrics Committee on Bioethics. 2001. Ethical issues with genetic testing in pediatrics. Pediatrics. 107:1452-1455. 2. American Academy of Pediatrics Committee on Genetics. 1989. Newborn screening fact sheets. Pediatrics. 83:449-464. 3. American Academy of Pediatrics Committee on Genetics. 2000. Molecular genetic testing in pediatric practice: A subject review. Pediatrics. 106:1494-1497. 4. Beutler E, Felitti VJ, Koziol JA, Ho NJ, Gelbart T. 2002. Penetrance of 845GA (C282Y) HFE hereditary haemochromatosis mutation in the USA. Lancet 359:211-218. 5. Bhardwaj U, Zhang Y-H, McCabe ERB. 2003. Neonatal hemoglobinopathy screening: Molecular genetic technologies. Mol Genet Metab. 80:129-137. 6. Burke W, Thomson E, Khoury MJ, McDonnell SM, Press N, et al. 1998. Herediatry homochromatosis: Gene discovery and its implications for population-based screening. JAMA. 280:172-178. 7. Chace DH. 2003. MMWR. 52:677. 8. Chace DH, DiPerna JC, Naylor EW. 1999. Laboratory integration and utilization of tandem mass spectrometry in neonatal screening: A model for clinical mass spectrometry in the next millennium. Acta Paed. 88:45-47. 9. Cohn ED, Kelley PM. 1999. Clinical phenotype and mutations in connexin 26 (DFNB1/GJB2), the most common cause of childhood hearing loss. Am J Med Genet. 89:130-136. 10. del Castillo I, Moreno-Pelayo MA, del Castillo FJ, Brownstein Z, Marlin S, Adina Q, et al. 2003. Prevalence and evolutionary origins of the del(GJB6-D13S1830) mutation in the DFNB1 locus in hearing-impaired subjects: A multicenter study. Am J Hum Genet. (Epub ahead of print) 11. del Castillo I, Villamar M, Moreno-Pelayo MA, del Castillo FJ, Alvarez A, Telleria D, et al. 2002. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. New Engl J Med. 346:243-9. 12. Dequecker E, Cassiman J-J. 2001. Quality evaluation of data interpretation and reporting. Am J Hum Genet. 69S:438. 13. Descartes M, Huang Y, Zhang Y-H, McCabe L, Gibbs R, Therrell BL Jr, McCabe ERB. 1992. Genotypic confirmation from original dried blood specimens in a neonatal hemoglobinopathy screening program. Ped Res. 31:217-221. 14. Gaston MH, Verter JI, Woods G, Pegelow C, Kelleher J, Presbury G, et al., and Prophylactic Penicillin Study Group. 1986. Prophylaxis with oral penicillin in children with sickle cell anemia- a randomized trial. N Engl J Med. 314:1593-1599. 15. Gau JJ, Lan EH, Dunn B, Ho, CM, Woo JC. 2001. A MEMS based amperometric detector for E. coli bacteria using self-assembled monolayers. Biosens Bioelecton. 16(912):745-55. 16. Government Accounting Office. 2003. Newborn Screening: Characteristics of State Programs. 17. Gregg RG, Simantel A, Farrell PM, Koscik R, Kosorok MR, et al. 1997. Newborn screening for cystic fibrosis in Wisconsin: Comparison of biochemical and molecular methods. Pediatrics. 99:819-824. 18. Grody WW, Cutting GR, Klinger KW, Richards CS, Watson MS, Desnick RJ. 2001. Laboratory standards and guidelines for population-based cystic fibrosis carrier screening. Genet Med. 3:149-154. 19. Grody WW, Desnick RJ. 2001. Cystic fibrosis population carrier screening: Here at last – are we ready? Genet Med. 3:87-90. 20. Grody WW, Griffin JH, Taylor AK, Korf BR, Heit JA. 2001. American College of Medical Genetics consensus statement on factor V Leiden mutation testing. Genet Med. 3:139-148. 21. Guthrie R, Susi A. 1963. A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics. 32:338-343. 22. Holtzman C, Slazyk WE, Cordero JF, Hannon WH. 1986. Descriptive epidemiology of missed cases of phenylketonuria and congenital hypothyroidism. Pediatrics. 78:553-558. 23. Insinga RP, Laessig RH, Hoffman GL. 2002. Newborn screening with tandem mass spectrometry: examining its cost-effectiveness in the Wisconsin Newborn Screening Panel. J Pediatr. 141(4):524-31. 24. Jinks DC, Minter M, Tarver DA, Vanderford M, Hejtmancik JF, McCabe ERB. 1989. Molecular genetic diagnosis of sickle cell disease using dried blood specimens on blotters used for newborn screening. Hum Genet. 81:363-366. 25. Khoury MJ, McCabe LL, McCabe ERB. 2003. Population screening in the age of genomic medicine. New Engl J Med. 348:50-58. 26. Koch JH. 1997. Robert Guthrie: The PKU Story, Pasadena, CA: Hope Publishing House. 27. Lehto M, Tuomi T, Mahtani MM, Widen E, Forsblom C, et al. 1997. Characterization of the MODY3 phenotype. Early-onset diabetes caused by an insulin secretion defect. J Clin Invest. 99:582-591. 28. Lehto M, Wipemo C, Ivarsson SA, Lindgren C, Lipsaren- Nyman M, et al. 1999. High frequency of mutations in MODY and mitochondrial genes in Scandanavian patients with familial early-onset diabetes. Diabetologia. 42:1131-1137. 29. Lewis MH, McCabe LL, and McCabe ERB. 2003. Newborn screening blood samples: A vulnerable DNA database. American Journal of Human Genetics. 73S:257. 30. Liu XZ, Xia XJ, Adams J, Chen ZY, Welch KO, Tekin M, et al. 2001. Mutations in GJA1 (connexin 43) are associated with non-syndromic autosomal recessive deafness. Hum Mol Genet. 10(25):2945-51. 31. Mak V, Zielenski J, Tsui L-C, Durie P, Zini A, et al. 1999. Proportion of cystic fibrosis gene mutations not detected by routine testing in men with obstructive asoospermia. JAMA. 281:2217-2224. 32. McCabe ERB. 2002. ACMG Presidential Address: Translational genomics in medical genetics. Genetics in Medicine. 4:468-471. 33. McCabe ERB. 1991. Utility of PCR for DNA analysis from dried blood spots on filter paper blotters. PCR Meth Appl. 1:99-106. 34. McCabe ERB, Huang S-Z, Seltzer WK, Law ML. 1987. DNA microextraction from dried blood spots on filter paper blotters: Potential applications to newborn screening. Hum Genet. 75:213-216. 35. McCabe ERB, McCabe L, Mosher GA, Allen RJ, Berman JL. 1983. Newborn screening for phenylketonuria: Predictive validity as a function of age. Pediatrics. 72:390-398. 36. McCabe L. 1996. Efficacy of a targeted genetic screening program for adolescents. Am J Hum Genet. 59:762-763. 37. McCabe LL, McCabe ERB. 2002. Newborn screening as a model for population screening. Mol Genet Metab. 75:299-307. 38. McCabe LL, Therrel BL Jr, McCabe ERB. 2002. Newborn screening: Rationale for a comprehensive, fully integrated public health system. Mol Genet Metab. 77:267-273. 39. Mitchell JJ, Capua A, Clow C, Scriver CR. 1996. Twenty-year outcome analysis of genetic screening programs for Tay-Sachs and beta-thalassemia disease carriers in high schools. Am J Hum Genet. 59:793-798. 40. Morrell RJ, Kim JH, Hood IJ, GoForth L, Friderici K, et al. 1998. Mutations in the connexin 26 gene (GJB2) among Ashkenazie Jews with nonsyndromic recessive deafness. New Engl J Med. 339:1500-1505. 41. National Academy of Sciences. 1975. Genetic Screening: Programs, Principles, and Research. Washington, DC. 42. National Institutes of Health Consensus Development Conference Statement on genetic testing for cystic fibrosis. 1999. Arch. Intern. Med. 159:1529-1539. 43. Naylor EW, Chace DH. 1999. Automated tandem mass spectrometry for mass newborn screening for disorders in fatty acid, organic acid, and amino acid metabolism. J Child Neurol. 14:S4-S8. 44. Newborn Screening Task Force Report. 2000. Serving the family from birth to the medical home. Pediatrics. 106S:386-427. 45. Raman V, Clary R, Siegrist KL, Zehnbauer B, Chatila TA. 2002. Increase prevalence of mutations in the cystic fibrosis transmembrane conductance retulator in children with chronic rhinosinusitis. Pediatrics. 109:136-137. 46. Rosenbloom AL, Joe JR, Young RS, Winter WE. 1999. Emerging epidemic of type 2 diabetes in youth. Diabetes Care. 22:345-354. 47. Rubin EM, Andrews KA, Kan YW. 1989. Newborn screening by DNA analysis of dried blood spots. Hum Genet. 82(2):134-136. 48. Schoen EJ, Baker JC, Colby CJ, To TT. 2002. Cost-benefit analysis of universal tandem mass spectrometry for newborn screening. Pediatrics. 110(4):781-6. 49. Schulze A, Lindner M, Kohlmuller D, Olgemoller K, Mayatepek E, Hoffmann GF. 2003. Expanded newborn screening for inborn errors of metabolism by electrospray ionization-tandem mass spectrometry: results, outcome, and implications. Pediatrics. 111:1399-406. 50. Scriver CR and Committee. 1985. Population screening: report of a workshop. Prevention of Physical and Mental Congenital Defects. Part B: Epidemiology, Early Detection and Therapy, and Environmental Factors. Alan R Liss Inc., New York. 51. Therrell BL Jr. 2001. U.S. newborn screening policy dilemmas for the twenty-first century. Mol Genet Metab. 74:64-74. 52. Vandenbroucke JP, Koster T, Briet E, Reitsma PH, Bertina RM, Rosendaal FR. 1994. Increased risk of venous thrombosis in oral-contraceptive users who are carriers of factor V Leiden mutation. Lancet. 344:1453-1457. 53. Vanderbroucke JP, van der Meer FJM, Helmerhorst FM, Rosendall FR. 1996. Factor V Leiden: Should we screen oral contraceptive users and pregnant women? BMJ. 313:1127-1130. 54. Wang XJ, Moylan B, Leopold DA, Kim J, Rubenstein RC, et al. 2000. Mutation in the gene responsible for cystic fibrosis and predisposition to chronic rhinosinusitis in the general population. JAMA. 284-1814-1819. 55. Weigl BH, Bardell RL, Cabrera CR. 2003. Lab-on-a-chip for drug development. Adv Drug Deliv Rev. 55(3):349-77. 56. Wilcken B, Wiley V, Hammond J, Carpenter K. 2003. Screening newborns for inborn errors of metabolism by tandem mass spectometry. New England Journal of Medicine. 348(23):2304-2312. 57. Wilcox RL, Nelson CC, Stenzel P, Steiner RD. 2002. Postmortem screening for fatty acid oxidation disorders by analysis of Guthrie cards with tandem mass spectrometry in sudden unexpected death in infancy. J Pediatr. 141(6):833-6. 58. Williams C, Weber L, Williamson R, Hjelm M (1988) Guthrie spots for DNA-based carrier testing in cystic fibrosis. Lancet II:693 59. Witte DL, Crosby WH, Edwards CQ, Fairbanks VF, Mitros FA. 1996. Practice guideline development task force of the College of American Pathologists. Hereditary hemochromatosis. Clin Chim Acta. 245:139-200. 60. Zhang Y-H, McCabe ERB. 1992. RNA analysis from newborn screening dried blood specimens. Hum Genet. 89:311-314. 61. Zhang Y-H, McCabe L, Wilborn M, Therrell BL Jr, McCabe ERB. 1994. Application of molecular genetics in public health: Improved follow-up in a neonatal hemoglobinopathy screening program. Biochem Med Metab Biol. 52:27-35.