Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
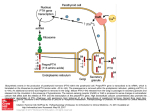
REVIEW Cardiovascular Research (2012) 94, 10–19 doi:10.1093/cvr/cvs092 Aldosterone and parathyroid hormone: a precarious couple for cardiovascular disease Andreas Tomaschitz 1*†, Eberhard Ritz 2, Burkert Pieske 1, Astrid Fahrleitner-Pammer 3, Katharina Kienreich 3, Jörg H. Horina 4, Christiane Drechsler 5, Winfried März 6,7,8, Michael Ofner 9, Thomas R. Pieber 3, and Stefan Pilz 3,10† 1 Division of Cardiology, Department of Internal Medicine, Medical University of Graz, Auenbruggerplatz 15, 8036 Graz, Austria; 2Division of Nephrology, Department of Medicine, University Hospital Heidelberg, Heidelberg, Germany; 3Division of Endocrinology and Metabolism, Department of Internal Medicine, Medical University of Graz, Graz, Austria; 4Division of Nephrology, Department of Internal Medicine, Medical University of Graz, Graz, Austria; 5Division of Nephrology, Department of Medicine, University of Würzburg, Würzburg, Germany; 6 Clinical Institute of Medical and Chemical Laboratory Diagnostics, Medical University of Graz, Graz, Austria; 7Synlab Academy, Synlab services LLC, Mannheim, Germany; 8Medical Faculty Mannheim, Mannheim Institute of Public Health, Ruperto Carola University Heidelberg, Mannheim, Germany; 9Frank Stronach Institute of Health, Oberwaltersdorf, Austria; and 10 Department of Epidemiology and Biostatistics and EMGO Institute for Health and Care Research, VU University Medical Center, Amsterdam, The Netherlands Received 12 October 2011; revised 2 February 2012; accepted 9 February 2012; online publish-ahead-of-print 13 February 2012 Abstract Animal and human studies support a clinically relevant interaction between aldosterone and parathyroid hormone (PTH) levels and suggest an impact of the interaction on cardiovascular (CV) health. This review focuses on mechanisms behind the bidirectional interactions between aldosterone and PTH and their potential impact on the CV system. There is evidence that PTH increases the secretion of aldosterone from the adrenals directly as well as indirectly by activating the renin–angiotensin system. Upregulation of aldosterone synthesis might contribute to the higher risk of arterial hypertension and of CV damage in patients with primary hyperparathyroidism. Furthermore, parathyroidectomy is followed by decreased blood pressure levels and reduced CV morbidity as well as lower renin and aldosterone levels. In chronic heart failure, the aldosterone activity is inappropriately elevated, causing salt retention; it has been argued that the resulting calcium wasting causes secondary hyperparathyroidism. The ensuing intracellular calcium overload and oxidative stress, caused by PTH and amplified by the relative aldosterone excess, may increase the risk of CV events. In the setting of primary aldosteronism, renal and faecal calcium loss triggers increased PTH secretion which in turn aggravates aldosterone secretion and CV damage. This sequence explains why adrenalectomy and blockade of the mineralocorticoid receptor tend to decrease PTH levels in patients with primary aldosteronism. In view of the reciprocal interaction between aldosterone and PTH and the potentially ensuing CV damage, studies are urgently needed to evaluate diagnostic and therapeutic strategies addressing the interaction between the two hormones. ----------------------------------------------------------------------------------------------------------------------------------------------------------Keywords Aldosterone † Parathyroid hormone † Cardiovascular disease 1. Introduction Research in cardiovascular (CV) endocrinology deals with vascular and myocardial pathology caused by dysregulated endocrine systems. The search for novel endocrine parameters to assess their potential role in the pathophysiology of CV complications is a novel challenge in endocrinology. In the past, dysregulation of aldosterone as well as of parathyroid hormone (PTH) has been recognized to play an important role in the development and progression of cardiovascular disease (CVD). Less is known, however, about the recently recognized reciprocal interaction between these two hormones and its potential role for target organ damage. Specifically, the mechanisms involved in the interaction between aldosterone and PTH are poorly understood and this issue is largely ignored in clinical routine. The delineation of this bidirectional interaction is hampered by multiple factors, such as the activity of the sympathetic nervous system, which impact on both renin –angiotenin–aldosterone system (RAAS) activation and PTH secretion, respectively.1,2 Due to the complex regulation of either hormone, it is beyond the scope of this review to consider all regulatory mechanisms in detail. Rather, we attempt to provide an overview specifically addressing the physiology and pathophysiology of the interaction between aldosterone * Corresponding author. Tel: +43 664 1443993; fax: +43 316 385 13733, Email: [email protected] † Both authors contributed equally to the manuscript. Published on behalf of the European Society of Cardiology. All rights reserved. & The Author 2012. For permissions please email: [email protected]. 11 Aldosterone and parathyroid hormone and PTH as well as its potential impact on CV health in three different scenarios: (i) primary hyperparathyroidism; (ii) chronic heart failure; and (iii) primary aldosteronism. 2. Physiology and pathophysiology of aldosterone and parathyroid hormone Accumulating evidence points to an eminent role of the mineralocorticoid hormone aldosterone, produced with the zona glomerulosa (ZG) of the adrenal gland, in the pathogenesis of CV and renal diseases.3 The renin –angiotensin system, potassium, and adrenocorticotropic hormone are major regulators of adrenal aldosterone synthesis. In its action, aldosterone binds to the mineralocorticoid receptor (MR) and regulates gene transcription of the epithelial sodium channel in endothelial cells and in the renal collecting duct, increasing vascular stiffness on the one hand and promoting sodium retention on the other hand.4,5 Given that the MR has also been identified in non-epithelial tissues, such as vascular smooth muscle cells as well as cardiomyocytes, the classic view that aldosterone acts exclusively on transport epithelial cells has been broadened to include cells other than transport epithelia.6 It is increasingly recognized that, even in the absence of primary aldosteronism, a relative excess of aldosterone, i.e. in the absence of aldosterone concentrations above the ‘normal range’—aldosterone may play an important role in the genesis of arterial hypertension and CV damage.7 – 9 The activated MR –aldosterone complex regulates the transcription of myriads of genes in a tissue-specific pattern.10 Experimental and clinical studies documented that aldosterone-mediated proinflammatory and profibrotic effects were associated with left ventricular hypertrophy and reduced kidney function.11 – 13 We and others recently reported a strong and independent association between plasma aldosterone levels within the ‘normal range’ and CV mortality, in particular fatal stroke and sudden cardiac death.14,15 PTH is secreted by the chief cells in the parathyroid gland mainly in response to a decreased circulating ionized calcium concentration.16 In addition, calcitriol, phosphate, magnesium, the FGF23/klotho system, and other factors participate in the regulation of PTH synthesis.17 PTH acts via binding to (i) the PTH/PTH-related protein receptor (PTH/PTH-rP receptor ¼ PTH1R), (ii) the NH2-terminal PTH receptor II (PTHR2), or (iii) the COOH terminal PTH receptor (C-PTHR).18,19 PTH is a crucial regulator of calcium and phosphate homeostasis. This goal is achieved by activating osteoclasts and osteoblasts, enhancing intestinal Ca2+ absorption, promoting the synthesis of active vitamin D in the kidney, and increasing active renal Ca2+ reabsorption. The subsequent elevation of plasma Ca2+ concentration in turn lowers PTH secretion by activating calcium sensing receptors located on chief cells. The close control of ionized calcium levels is essential for the maintenance of a plethora of processes, such as cell signalling, neuromuscular function, and bone metabolism. The identification of PTH receptors within the CV system, for example, in cardiomyocytes, vascular smooth muscle, and endothelial cells, indicates that PTH excess may have potential effects beyond the regulation of calcium and phosphate homeostasis.20 3. The interplay between aldosterone and parathyroid hormone in primary hyperparathyroidism Primary hyperparathyroidism, the third most common endocrine disorder, is characterized by excess PTH secretion, i.e. secretion inappropriate with respect to the prevailing concentration of ionized calcium. Most patients with primary hyperparathyroidism have no characteristic symptoms; in the majority of cases, excess PTH concentrations are detected incidentally. In the long term, primary hyperparathyroidism is associated with the development of osteoporosis and the ensuing fracture risk. Although this had not been realized in the distant past, patients with primary hyperparathyroidism have a remarkably higher risk to die from CV causes compared with the general population.21,22 In addition, various observational studies linked elevated PTH levels to a higher risk of hypertension, left ventricular hypertrophy, arrhythmia, diabetes, hyperlipidaemia, and, most importantly, CV morbidity and mortality.23 – 26 Furthermore, in patients with CVD and in elderly men, prospective studies revealed strong and independent associations between higher PTH levels and increased CV mortality.27,28 Even a minor asymptomatic PTH excess is associated with a higher risks of all-cause mortality, fatal, and non-fatal CVD as well as of renal failure and renal stones.29 The interplay between PTH and aldosterone is increasingly suggested as an important mechanism underlying the increased risk of CV damage observed in primary hyperparathyroidism.30 Early evidence for a physiological and pathophysiological bidirectional link between aldosterone and PTH in humans had initially been derived mainly from case reports.31 To explain the biochemical changes following parathyroid surgery, it has been suggested that hyperaldosteronism might be caused (directly or indirectly) by primary hyperparathyroidism and vice versa.32,33 Several studies evaluated the RAAS after parathyroidectomy in animals and in humans with PTH excess. In 5/6 nephrectomized rats, a significant increase in aldosterone levels was observed compared with control rats. After combined 5/6 nephrectomy and parathyroidectomy, aldosterone levels were lower, but potentially still inappropriate, compared with control rats.34 Studies in patients with primary hyperparathyroidism documented markedly decreased plasma aldosterone levels and plasma renin activity after parathyroidectomy.35 – 37 Recently, Brunaud et al. 38 reported significantly decreased aldosterone and blood pressure levels after parathyroidectomy in 134 patients with primary hyperparathyroidism. In the majority of subsequent studies on primary hyperparathyroidism patients, a significant decline of plasma renin activity, of angiotensin II, and of aldosterone levels was documented after parathyroidectomy.39 – 42 Unfortunately, firm statements about the change of the RAAS components in the circulation after parathyroid surgery are precluded in view of the lack of the standardization of laboratory measurement of the circulating components of the RAAS, of the failure to consider the impact of blood pressure levels on RAAS activity per se, and in view of the small sample size of many studies.43 The pathophysiological background of the high prevalence of arterial hypertension, arterial stiffness, and CVD found in patients with elevated PTH levels is an important research issue with high clinical relevance.44 Several cross-sectional and prospective studies 12 documented a strong relationship between aldosterone levels and arterial hypertension as well as increased arterial stiffness.25,26,45 In view of the interaction between aldosterone and PTH, one might speculate that the interplay between both hormones aggravates blood pressure elevation, remodelling of blood vessels, and CVD in patients with elevated PTH.46 This hypothesis would be in line with the observation of Morfis et al.:25 aldosterone levels were strongly related to 24 h ambulatory blood pressure, but the correlation was less significant when the confounding effect of PTH was taken into consideration. This is all the more plausible because in healthy adults, continuous PTH infusion increased urinary tetrahydroaldosterone and blood pressure values.47 In patients with primary hyperparathyroidism, compared with healthy controls, blood pressure declined significantly 3 months after parathyroidectomy.48 In 16 patients with primary hyperparathyroidism, the decline in blood pressure after parathyroidectomy was accompanied by a parallel decrease in aldosterone levels.35 In one presumably underpowered study, a trend of reduced systolic blood pressure was noted after parathyroid surgery.37 A recent study assessed 134 primary hyperparathyroidism patients with arterial hypertension and/or a positive history of coronary artery disease; significantly higher aldosterone levels were found compared with normotensive individuals and probands without known coronary artery disease.38 Preoperative serum aldosterone levels were significantly higher in patients with PTH . 127 ng/L compared with those with PTH , 127 ng/L (P ¼ 0.019) independent of ongoing antihypertensive medication. In contrast, 3 months after surgery, no significant correlation was observed any longer between postoperative PTH and aldosterone levels. Although causality is not strictly proven, the current evidence supports the notion that after parathyroid surgery the lower blood pressure values and the cardio-/vasculoprotective effects are the result of less RAAS activation following the decrease in PTH. In our opinion, the above-mentioned evidence for a functional link between aldosterone and PTH justifies further mechanistic and interventional studies in order to evaluate the presumed beneficial effects of the MR-blockade on both CV health and rates of PTH secretion in patients with hyperparathyroidism. 3.1 Mechanisms underlying the functional interplay between aldosterone and parathyroid hormone Several experimental studies aimed to delineate the mechanisms underlying the effect of PTH on aldosterone secretion from the adrenals. Importantly, PTH stimulates the entry of cytosolic calcium (Ca2+) into the mitochondrial matrix and this step is essential for the initiation of steroidogenesis within the mitochondria.49 – 51 L-(high-threshold, long lasting), N-(neural) type, and T-type (lowthreshold, transient) voltage-gated calcium channels are essential for the control of the cellular calcium messenger system and have been identified in bovine and human ZG cells.52 – 54 Extracellular potassium and angiotensin II interact with voltage-gated calcium channels to depolarize the ZG cells causing a sustained calcium influx. After dissecting mitochondrial and cytosolic Ca2+ signals, Wiederkehr et al.55 recently demonstrated that matrix Ca2+ participates in the regulation of energy metabolism and of NAD(P)H concentrations in ZG cells, thus stimulating aldosterone synthesis. The Ca2+ messenger system further participates in the initiation of steroidogenesis by enhancing intramitochondrial cholesterol transfer into the mitochondria.56 In the setting of secondary hyperparathyroidism, calcium extrusion A. Tomaschitz et al. might be impaired causing elevated intracellular calcium levels in ZG cells.57 This finding is in line with the observation that angiotensin II maintains intracellular calcium levels by reducing calcium extrusion through activating the Na+/Ca2+ exchanger in ZG cells.58 In contrast, under physiological conditions, atrial natriuretic peptide reduces aldosterone secretion by inhibition of T-type calcium channels.59 It is still under investigation whether PTH stimulates adrenal aldosterone synthesis directly. Activation of both the PTH/PTH-rP receptor and voltage-gated L-type calcium channels mediates PTH-dependent calcium entry in various cell types.60,61 The PTH/ PTH-rP receptor which has also been identified in human and rat adrenal cortex binds intact PTH and the biologically active N-terminal fragment PTH 1–34.62,63 In various cell types, binding to the PTH1R activates multiple cellular signalling pathways, including cAMP, phospholipase C, protein kinase C, and, importantly, release of Ca2+ from intracellular calcium stores. For instance, Klin et al.64 noted that PTH-related calcium entry is receptor-mediated and involves the G protein-adenylate cyclase-cAMP system, activation of L-type calcium channels, and protein kinase C. Mazzocchi et al.65 and others demonstrated that in human adrenals, PTH and PTH-related protein increase aldosterone production by binding to the PTH/PTH-rP receptor, activating cellular adenylate cyclase/cAMPdependent protein kinase, phospholipase C/protein kinase C- and cAMP-dependent signalling cascades.66 Mechanistic studies attempted to shed light on the interplay between aldosterone and PTH by investigating the effects of PTH on RAAS activity and on aldosterone secretion from adrenal ZG cells, respectively. Olgaard et al.67 evaluated the effect of PTH on Ca2+-mediated aldosterone secretion in isolated rat ZG cells. Aldosterone release increased significantly by up to 200% above baseline values in cells exposed to PTH(1 –84) and PTH(1 –34). The authors suggested that PTH exerts Ca2+ ionophore-like effects in the ZG causing increased Ca2+-stimulated aldosterone secretion. One previous investigation had shown that in bovine ZG cells PTH alone induced only a slight increase in intracellular Ca2+, while the intracellular Ca2+ response was more pronounced after stimulation with angiotensin II.68 In patients with primary hyperparathyroidism, Fallo et al.69 compared the response of aldosterone to angiotensin II infusion before and after parathyroidectomy. Plasma aldosterone and renin activity did not vary significantly before and after the parathyroidectomy. In contrast in the hyperparathyroid patients, the aldosterone response to angiotensin II infusion was significantly greater than in healthy controls and more pronounced before than after surgery. The authors concluded that in hyperparathyroid patients, high levels of extracellular calcium or PTH, or both, play a major role in the exaggerated aldosterone response to angiotensin II. In healthy subjects, as well continuous (12 days) i.v. PTH infusion increased urinary tetrahydroaldosterone excretion significantly in parallel with the development of hypercalcaemia and hypertension.47 In healthy adults, Grant et al.70 observed an increase in plasma renin activity after PTH(1 –34) infusion without any change of ionized serum calcium concentration. Because, in addition, plasma cortisol levels were elevated after PTH infusion, Hulter et al.47 suggested that a transient calcium-mediated rise of adrenocorticotropic hormone had increased the secretion of adrenal steroid hormones. This would be in line with findings in experimental rat models, indicating that human PTH(1– 34) directly stimulates adrenal steroidogenesis, presumably by interacting with the receptor for adrenocorticotropic hormone 1– 39.71 Thus, currently available evidence derived from these mechanistic studies Aldosterone and parathyroid hormone is compatible with the assumption that both in patients with and without primary hyperparathyroidism, there is an interplay between aldosterone and PTH. We recently analysed the relation between PTH and plasma aldosterone concentration in 3296 patients enrolled in the Ludwigshafen Risk and Cardiovascular Health (LURIC) study who were referred to coronary angiography. We found a significant association between plasma aldosterone and plasma PTH levels, particularly in vitamin D insufficient patients.27,72 Considering, however, that vitamin D may suppress renal renin synthesis, we cannot exclude the possibility that in patients with vitamin D deficiency, elevated renin levels stimulate aldosterone secretion independent of PTH. These suggestions could explain the observation of Ozata et al.73 who found no association between aldosterone and PTH in male obese subjects; nevertheless in this patient group, upright plasma renin activity was correlated to PTH. In summary, the reported experimental and clinical data support the notion that PTH might stimulate adrenal aldosterone synthesis, both directly (by facilitating calcium entry into adrenal ZG cells via binding to PTH/PTH-rP receptor, voltage-gated L-type calcium channels, and adrenocorticotropic hormone-receptors) and indirectly (by stimulating renal renin release and increasing angiotensin II concentration—thus sensitizing adrenal ZG cells). Such stimulatory effects of PTH on the RAAS may potentiate the risks of development and progression of arterial hypertension as well as the risk of CVD in patients with primary hyperparathyroidism.47 Figure 1 summarizes the suggested pathways of the interplay between PTH and the RAAS in the setting of PTH excess. In epithelial tissues, activation of the MR by cortisol is mainly prevented by the cortisol-inactivating enzyme 11b-hydroxysteroid dehydrogenase-2. In the setting of increased generation of reactive oxygen species, e.g. in chronic kidney disease and heart failure, cortisol might also activate the MR—in addition to aldosterone—thus aggravating profibrotic and proinflammatory effects.4,74 To date it is unclear, however, whether cortisol affects renal handling of calcium via binding to the MR. One recent study revealed an upregulated expression of PTH-related peptide in the mice kidney after 4 weeks treatment with cortisol.75 In the past, only few clinical studies had addressed the relation between PTH and cortisol. In a small study of patients with primary hyperparathyroidism, circulating cortisol levels decreased significantly after parathyroidectomy.41 Conversely, intravenous infusion of PTH in healthy adults increased plasma cortisol concentration.47 Considering (i) that hypercalcaemia, caused by PTH excess, results in a transient rise of adrenocorticotropic hormone secretion; (ii) that PTH stimulates steroid hormone synthesis in part by binding to the adrenocorticotropic hormone receptor; and (iii) that cortisol upregulates PTH-related peptide, one might speculate that this sequence impacts on CV health. In view of the higher CVD risk in patients with hypercortisolism, the conceivable relationship between glucocorticoids and PTH should be addressed in further studies. 4. The interplay between aldosterone and parathyroid hormone in chronic heart failure The European Society of Cardiology estimates that the prevalence of HF in the population is around 4% and even 10–20% in people above age of 70 years; every second patient suffering from HF will die within 13 4 years.76 Neurohormonal activation is a hallmark of chronic HF.77 Low perfusion pressure due to impaired left ventricular function results in the activation of the hypothalamic-pituitary-adrenal axis and of the sympathetic nervous system. Stimulation of adrenal aldosterone synthesis in chronic HF occurs despite sodium and fluid retention. Impaired homeostasis of cations is frequent in patients with HF, resulting from the combination of a hyperadrenergic state (leading to translocation of cations into the intracellular compartment) with an increased aldosterone secretion (stimulating of faecal and urinary loss of cations), respectively.78 The resulting hypocalcaemia and hypomagnesaemia stimulate PTH secretion which tends to restore extracellular calcium and magnesium homeostasis. On the other hand, the PTH-promoted mitochondrial Ca2+ excess, e.g. in the myocardium, induces oxidative stress and necrotic cell death which in the long term causes or amplifies myocardial fibrosis aggravating systolic and diastolic HF. Importantly, as discussed above, PTH tends to further stimulate adrenal aldosterone synthesis, thus triggering a vicious circle of mutually reinforcing aldosteronism and hyperparathyroidisms with the resulting risk of even more target organ damage. Relative aldosterone excess causes sodium retention and oxidative stress, thus increasing CV morbidity and mortality.79 Conversely, inhibition of the MR improves survival in patients with different forms of HF: severe HF, HF after myocardial infarction, and even HF with mild symptoms.80 – 85 Important insight into the relationship between inappropriately elevated aldosterone and PTH levels has been gained by experimental studies performed by Weber et al.86 – 88 In rats, administration of aldosterone and 1% NaCl caused increased urinary and faecal Ca2+ and Mg2+ excretion, hypocalcaemia, hypomagnesaemia, and consequently secondary hyperparathyroidism as well as increased tissue calcium concentration. Moreover, as a result of increased PTH activity, bone mineral density and strength were significantly reduced. The role of aldosterone is underlined by the finding that urinary and faecal Ca2+ and Mg2+ excretion was attenuated by spironolactone. Furthermore, MR blockade improved bone mineral density and strength, reduced the intracellular calcium overload, and improved the redox status in peripheral blood mononuclear cells.89 Weber et al. suggested that as a result of the aldosterone-PTH interplay, the disequilibrium between the pro-oxidant calcium and the antioxidant zinc is a crucial factor in the pathogenesis of cardiomyocyte necrosis and myocardial fibrosis in chronic HF.90 In various cell types, elevated PTH further stimulates calcium influx by different pathways.91 The subsequent intracellular and mitochondrial calcium overload causes a disturbed redox status and increased oxidative stress in various tissues, i.e. in cardiac myocytes.90,92,93 In particular, when mitochondria are exposed to calcium overload and oxidative stress, sustained opening of the mitochondrial permeability transition pore is seen.94 This leads to reduction in intra-mitochondrial ATP levels and subsequent necrotic cell death and myocardial fibrosis.95 Presumably, these mechanisms explain, at least in part, the relationship between circulating aldosterone and PTH levels as well as the higher risk of left ventricular hypertrophy and sudden cardiac death.13,79 Increased aldosterone-mediated renal calcium loss might be the key mechanism for the subsequent development of hyperparathyroidism in chronic HF. The majority of studies demonstrated calcium wasting triggered by aldosterone, particularly in the setting of dietary salt excess, although conflicting results were reported.96 – 99 In an attempt to counteract calcium loss, the resulting hypocalcaemia and hypomagnesaemia triggers secondary hyperparathyroidism. 14 A. Tomaschitz et al. Figure 1 Suggested pathways of the interplay between PTH and the renin – angiotensin – aldosterone system in the setting of PTH excess. Abbreviations: BMD, bone mineral density; PTH (rP), parathyroid hormone (related peptide); ACTH, adrenocorticotropic hormone; ANG II, angiotensin II; ZG, zona glomerulosa; JG, juxtaglomerular; MR, mineralocorticoid receptor; ACE, angiotensin concerting enzymes; AT1-receptor, angiotensin II type 1 receptor. PTH (excess) increases circulating ionized Ca2+ (via increasing Ca2+ release from bone and decreasing renal Ca2+ excretion). PTH is suggested to stimulate renin synthesis by increasing calcium levels in JG cells. Renal renin synthesis is further controlled by tubular sodium concentration, arterial blood pressure, and the sympathetic nervous system. Extracellular potassium and angiotensin II are major stimulators of aldosterone synthesis in the adrenal glands. Both factors interact with voltage-gated calcium channels and depolarize the ZG cells which result in elevated intracellular calcium levels. PTH might also directly stimulate aldosterone synthesis by binding to the PTH/PTH-rP receptor, voltage-gated calcium channels, and the adrenocorticotropic hormone receptor, which results in increased mitochondrial Ca2+ levels. In addition, PTH is suggested to increase sensitization towards angiotensin II which by itself reduces cellular calcium extrusion through activating Na+/Ca2+ exchangers in ZG cells. PTH contributes to the development of arterial stiffness, arterial hypertension, and cardiac hypertrophy via binding to the PTH/PTH-rP receptor, which is expressed in vascular smooth muscle cells and cardiomyocytes. In addition, aldosterone, i.e. relative aldosterone excess, exerts genomic (by binding to the MR), and non-genomic profibrotic and proinflammatory effects on blood vessels and the myocardium. This may explain that increased levels of PTH, i.e. secondary hyperparathyroidism, are found in many patients with severe chronic HF.100 Furthermore, salt loading may also increase renal calcium elimination independent of aldosterone. To date, it is unclear whether relative aldosterone excess causes calcium wasting even in the absence of dietary salt excess. Weber et al. suggested that decreased reabsorption of Na+, Mg2+, and Ca2+ in the distal tubule, caused by salt retention and volume expansion, is responsible for aldosterone driven excretion of calcium and magnesium.89 Rossi et al.101 demonstrated that the effect of salt loading on renal calcium loss was even more pronounced in patients with primary aldosteronism compared with patients with essential hypertension. Aldosterone itself may cause intracellular calcium excess by upregulating T-type (lowthreshold, transient) calcium channels in various cell types.102 In aldosterone salt-treated rats, Vidal et al. documented an altered redox state, reflected by decreased levels of a1-antiproteinase.103 Importantly, oxidative stress in this experimental setting is attenuated by calcium and magnesium supplementation.104 Despite the recent decline in risk-adjusted HF hospitalization and risk-adjusted 1-year mortality rates between 1998 and 2008 in the USA, the 1-year overall mortality within heart failure patients remains unacceptably high.105 Despite a class I recommendation for 15 Aldosterone and parathyroid hormone their use in heart failure (NYHA class III/IV), MR blockers are still underused in this patient group.106,107 Given the increasing evidence that the potential interplay between aldosterone and PTH might contribute to the pathogenesis of HF and CVD, it should be determined whether more consistent use of MR blockers improves outcomes in CV risk patients; certainly contraindications must be considered and regular monitoring for side effects is mandatory.108 5. The interplay between aldosterone and parathyroid hormone in primary aldosteronism Primary aldosteronism, i.e. an absolute excess of aldosterone, is characterized by excessive adrenal aldosterone secretion out of proportion to its principal stimulant renin. The estimated prevalence of primary aldosteronism is 5–12% in arterial hypertension and 17– 23% in drug-resistant hypertension.109 Absolute aldosterone excess is strongly associated with a higher risk of development and progression of left ventricular hypertrophy, coronary artery disease, sudden cardiac death, chronic kidney disease, and strokes.110 – 112 Likewise in chronic HF hypercalciuria, hypocalcaemia and secondary hyperparathyroidism with subsequent intracellular calcium overload is frequently found in patients with low-renin hypertension and primary aldosteronism.113 – 115 Resnick et al.113 suggested that the interplay between the hormones regulating calcium homeostasis and the RAAS might contribute to the pathogenesis of arterial hypertension, particularly salt-sensitive hypertension. They also noted remarkable elevation of PTH levels in the majority of patients with primary aldosteronism.116 After adrenalectomy, a marked increase in ionized calcium concentration was observed. Furthermore, it has been suspected that patients with primary aldosteronism are at higher risk of developing renal calculi as a result of increased calcium excretion due to the calciuretic effect of aldosterone excess.117 In addition, in this setting, hypocitraturia was caused by aldosterone.118 These data led to the hypothesis that calcium intake and blockade of calcium channels might attenuate the aldosterone-PTH driven cascade of intracellular calcium overload and the resulting organ damage.119 This concept is in line with the finding of preserved bone integrity by co-treatment of hydrochlorothiazide plus spironolactone in aldosterone-salt-treated rats.86 So far, only few studies indicated that hyperparathyroidism is a common feature in primary aldosteronism. Nevertheless, Rossi et al.120 observed significantly higher serum concentrations of intact PTH in patients with PA compared with patients with essential hypertension. After 1 month of MR blockade with 100 mg spironolactone daily, an increase in serum-ionized calcium and a decrease in PTH level was observed. Recently, we compared the effects of MR blockade and adrenalectomy on PTH levels in patients with primary aldosteronism enrolled in the Graz Endocrine Causes of Hypertension (GECOH) study.121 In participants with primary aldosteronism, significantly higher PTH levels were found compared with those participants with essential hypertension.122 A non-significant trend of higher calcium-creatinine ratios was found in patients with primary aldosteronism. These patients also had significantly lower serum calcium levels compared with patients with essential hypertension. This finding supports the results of animal studies documenting increased aldosterone driven renal calcium loss and ensuing secondary hyperparathyroidism. Both regimens, i.e. adrenal surgery and treatment with MR blockers, were associated with a decline of PTH and arterial blood pressure during follow-up; this finding was not explained by changes in vitamin D status. A recent report supports our findings: compared with patients with essential hypertension, significantly higher PTH levels were found in patients with aldosteroneproducing adenomas, and again no difference in vitamin D status was seen.123 Importantly, adrenalectomy was followed by a non-significant decrease in the urinary calcium excretion. More studies are needed to confirm that lower serum calcium levels in patients with primary aldosteronism are mainly due to aldosterone-induced renal calcium loss. After adrenalectomy, however, normalization of PTH levels as well as an increase in serum-ionized calcium concentration was observed. Interestingly, the authors measured the expression of PTH/PTH-rP receptor on aldosterone-producing adenoma cells, underlining PTH-mediated effects on aldosterone-producing cells. Finally, whether PTH/PTH-rP receptor activation enhances tumour growth of aldosterone-producing adenomas, as shown in H295R adrenocortical tumour cells, remains to be determined.124 Collectively, these observations support the possibility of a clinically relevant interaction between aldosterone and PTH, presumably potentiating the CV risk in patients with primary aldosteronism. The mechanisms behind this link remain elusive. Such mechanisms are not necessarily similar to those which increase PTH in secondary hyperaldosteronism, e.g. in patients with chronic HF. A future task is the evaluation whether the measurement of PTH and the inhibition of PTH-mediated effects have implications for the diagnostic work-up and outcome of patients with aldosterone excess. Figure 2 gives an overview of the CV impact caused by the interaction of aldosterone and PTH in patients with chronic HF and aldosterone excess. 6. Summary and perspectives The majority of experimental animal studies and studies in humans support a clinically relevant interplay between aldosterone and PTH levels. It has been suggested that treatment of either disease, aldosterone excess, or hyperparathyroidism might positively affect the CV system by decreasing the activity of either hormone. The reduction in circulating aldosterone concentrations and in parallel of systolic/diastolic blood pressure values observed after parathyroidectomy suggests that the protective effect of surgery may be mediated, at least in part by reduction in RAS activity and aldosterone synthesis. Conversely, adrenalectomy or MR blockers, both decrease PTH secretion, arterial blood pressure as well as bone resorption. The novel perception of a functional link between aldosterone and PTH might be one more pathway of RAAS-mediated organ damage. This finding should encourage the development of novel treatment strategies to prevent CV disease.125 – 127 The emergence of novel CV risk factor constellations, e.g. the interactions between PTH, aldosterone, and renin, provide encouraging perspectives for the diagnosis and for the individually tailored treatment of patients at risk.27,79,128,129 Considering the fact that suboptimal blood pressure control globally accounted for tremendous health costs, it would be valuable both economically and for understanding the pathophysiology to measure circulating levels of aldosterone, renin, and PTH in patient of CV risk.130 Much must be learned about hormone synthesis, secretion, and elimination rates and about the interaction between hormones and receptors in target tissues. For example, recent evidence points to an important role of central haemodynamic effects of 16 A. Tomaschitz et al. Figure 2 Overview of the CV impact caused by the interaction of aldosterone and PTH in patients with chronic HF and aldosterone excess. BMD, bone mineral density; PTH (rP), parathyroid hormone (related peptide); Ang II, angiotensin II; MR, mineralocorticoid receptor. Activation of the renin – angiotensin – aldosterone system in the setting of heart failure results in salt/water retention and urinary loss of cations. Elevated PTH stimulate renal renin and adrenal aldosterone synthesis. Elevated aldosterone levels in primary and secondary hyperaldosteronism are paralleled by increased urinary and faecal loss of magnesium and calcium. The resulting lowering of serum calcium concentration further stimulates production of PTH which in turn amplifies adrenal aldosterone synthesis. PTH excess in turn induces calcium overload and oxidative stress in cardiomyocytes and aggravates the reduction in intra-mitochondrial ATP levels resulting in subsequent necrotic cell death and myocardial fibrosis. Finally, the vicious circle between aldosterone and PTH might potentiate CV damage. mineralocorticoids in mediating salt-dependent blood pressure elevation. Activation of the MRs, which are expressed in the circumventricular organs and amygdale, increases salt appetite, endogenous ouabain release, arginine vasopressin release, and sympathetic nervous system activity.131,132 Apart from the central nervous system, the functional link between aldosterone and PTH might be modified by molecular defects of hormone synthesis, signalling, e.g. receptor abnormalities and responsiveness.133 Genetic and epigenetic approaches are warranted to evaluate the biological pathways underlying inter-individual variation in blood pressure and CV risk and the responsiveness of complex endocrine systems on environmental stimuli.134,135 It remains to be determined whether blocking aldosterone-induced MR activation is organ protective by inhibiting the calcium wasting properties of aldosterone and subsequent PTH secretion. In addition, novel insight into the functional link between aldosterone and PTH in primary and secondary aldosteronism might be generated by the upcoming PTHR blockers.136 Further studies should therefore evaluate (i) the mechanisms behind the interplay between aldosterone and PTH, (ii) whether this interplay potentiates CV damage, and (iii) whether MR blockade breaks the vicious circle of the interdependence of aldosterone and PTH in various CV risk groups. Acknowledgements The authors thank Ms Tanja Traussnigg (www.mika-design.at) and Ms Dunja Bacinger Tomaschitz for providing the artwork of this manuscript. Conflict of interest: none declared. Funding K.K. is supported by funding from the Austrian National Bank (Jubilaeumsfond: project numbers: 13905 and 13878). This work was supported by the EU Project “MASCARA” (“Markers for Sub-Clinical Cardiovascular Risk Assessment”; THEME HEALTH.2011.2.4.2-2; Grant agreement no: 278249, BioPersMed (COMET K-project 825329), which is funded by Aldosterone and parathyroid hormone the Federal Ministry of Transport, Innovation and Technology (BMVIT), the Federal Ministry of Economics and Labour/the Federal Ministry of Economy, Family and Youth (BMWA/BMWFJ), and the Styrian Business Promotion Agency (SFG). References 1. Persson AE, Ollerstam A, Liu R, Brown R. Mechanisms for macula densa cell release of renin. Acta Physiol Scand 2004;181:471 – 474. 2. Schmitt CP, Obry J, Feneberg R, Veldhuis JD, Mehls O, Ritz E et al. Beta1-adrenergic blockade augments pulsatile PTH secretion in humans. J Am Soc Nephrol 2003;14: 3245 –3250. 3. Briet M, Schiffrin EL. Aldosterone: effects on the kidney and cardiovascular system. Nat Rev Nephrol 2010;6:261 –273. 4. Funder JW, Mihailidou AS. Aldosterone and mineralocorticoid receptors: clinical studies and basic biology. Mol Cell Endocrinol 2009;301:2–6. 5. Oberleithner H, Riethmuller C, Schillers H, MacGregor GA, de Wardener HE, Hausberg M. Plasma sodium stiffens vascular endothelium and reduces nitric oxide release. Proc Natl Acad Sci USA 2007;104:16281 –16286. 6. Connell JM, MacKenzie SM, Freel EM, Fraser R, Davies E. A lifetime of aldosterone excess: long-term consequences of altered regulation of aldosterone production for cardiovascular function. Endocr Rev 2008;29:133 –154. 7. Tomaschitz A, Pilz S, März W. Arterial hypertension and cardiovascular disease— absolute aldosterone excess is the tip of the iceberg. J Lab Med 2011;35:147 –151. 8. Vasan RS, Evans JC, Larson MG, Wilson PW, Meigs JB, Rifai N et al. Serum aldosterone and the incidence of hypertension in nonhypertensive persons. N Engl J Med 2004;351:33–41. 9. Tomaschitz A, März W, Pilz S, Ritz E, Scharnagl H, Renner W et al. Aldosterone/ renin ratio determines peripheral and central blood pressure values over a broad range. J Am Coll Cardiol 2010;55:2171 –2180. 10. Viengchareun S, Le Menuet D, Martinerie L, Munier M, Pascual-Le Tallec L, Lombes M. The mineralocorticoid receptor: insights into its molecular and (patho)physiological biology. Nucl Recept Signal 2007;5:e012. 11. Brilla CG, Weber KT. Mineralocorticoid excess, dietary sodium, and myocardial fibrosis. J Lab Clin Med 1992;120:893 –901. 12. Rocha R, Chander PN, Zuckerman A, Stier CT Jr. Role of aldosterone in renal vascular injury in stroke-prone hypertensive rats. Hypertension 1999;33:232 –237. 13. Edelmann F, Tomaschitz A, Wachter R, Gelbrich G, Knoke M, Dungen HD et al. Serum aldosterone and its relationship to left ventricular structure and geometry in patients with preserved left ventricular ejection fraction. Eur Heart J 2012;33: 203 –212. 14. Tomaschitz A, Pilz S, Ritz E, Grammer T, Drechsler C, Boehm BO et al. Association of plasma aldosterone with cardiovascular mortality in patients with low estimated GFR: the Ludwigshafen Risk and Cardiovascular Health (LURIC) Study. Am J Kidney Dis 2011;57:403 –414. 15. Ivanes F, Susen S, Mouquet F, Pigny P, Cuilleret F, Sautiere K et al. Aldosterone, mortality, and acute ischaemic events in coronary artery disease patients outside the setting of acute myocardial infarction or heart failure. Eur Heart J 2012;33:191 –202. 16. Murray TM, Rao LG, Divieti P, Bringhurst FR. Parathyroid hormone secretion and action: evidence for discrete receptors for the carboxyl-terminal region and related biological actions of carboxyl- terminal ligands. Endocr Rev 2005;26:78–113. 17. Silver J, Naveh-Many T. FGF23 and the parathyroid glands. Pediatr Nephrol 2010;25: 2241 –2245. 18. Clemens TL, Cormier S, Eichinger A, Endlich K, Fiaschi-Taesch N, Fischer E et al. Parathyroid hormone-related protein and its receptors: nuclear functions and roles in the renal and cardiovascular systems, the placental trophoblasts and the pancreatic islets. Br J Pharmacol 2001;134:1113 –1136. 19. Behar V, Pines M, Nakamoto C, Greenberg Z, Bisello A, Stueckle SM et al. The human PTH2 receptor: binding and signal transduction properties of the stably expressed recombinant receptor. Endocrinology 1996;137:2748 –2757. 20. Monego G, Arena V, Pasquini S, Stigliano E, Fiaccavento R, Leone O et al. Ischemic injury activates PTHrP and PTH1R expression in human ventricular cardiomyocytes. Basic Res Cardiol 2009;104:427–434. 21. Silverberg SJ, Lewiecki EM, Mosekilde L, Peacock M, Rubin MR. Presentation of asymptomatic primary hyperparathyroidism: proceedings of the third international workshop. J Clin Endocrinol Metab 2009;94:351 –365. 22. Ljunghall S, Jakobsson S, Joborn C, Palmer M, Rastad J, Akerstrom G. Longitudinal studies of mild primary hyperparathyroidism. J Bone Miner Res 1991;6(Suppl. 2): S111 –S116; discussion S121 –114. 23. Kamycheva E, Sundsfjord J, Jorde R. Serum parathyroid hormone level is associated with body mass index. The 5th Tromso study. Eur J Endocrinol 2004;151:167 – 172. 24. Nilsson IL, Aberg J, Rastad J, Lind L. Left ventricular systolic and diastolic function and exercise testing in primary hyperparathyroidism-effects of parathyroidectomy. Surgery 2000;128:895–902. 25. Morfis L, Smerdely P, Howes LG. Relationship between serum parathyroid hormone levels in the elderly and 24 h ambulatory blood pressures. J Hypertens 1997;15: 1271 –1276. 17 26. Rubin MR, Maurer MS, McMahon DJ, Bilezikian JP, Silverberg SJ. Arterial stiffness in mild primary hyperparathyroidism. J Clin Endocrinol Metab 2005;90:3326 –3330. 27. Pilz S, Tomaschitz A, Drechsler C, Ritz E, Boehm BO, Grammer TB et al. Parathyroid hormone level is associated with mortality and cardiovascular events in patients undergoing coronary angiography. Eur Heart J 2010;31:1591 –1598. 28. Hagstrom E, Hellman P, Larsson TE, Ingelsson E, Berglund L, Sundstrom J et al. Plasma parathyroid hormone and the risk of cardiovascular mortality in the community. Circulation 2009;119:2765 –2771. 29. Yu N, Donnan PT, Leese GP. A record linkage study of outcomes in patients with mild primary hyperparathyroidism: The Parathyroid Epidemiology and Audit Research Study (PEARS). Clin Endocrinol (Oxf) 2011;75:169 – 176. 30. Pilz S, Tomaschitz A, März W, Cavalier E, Ritz E. Aldosterone and parathyroid hormone: a complex and clinically relevant relationship. Calcif Tissue Int 2010;87: 373– 374. 31. Barkan A, Marilus R, Winkelsberg G, Yeshurun D, Blum I. Primary hyperparathyroidism: possible cause of primary hyperaldosteronism in a 60-year-old woman. J Clin Endocrinol Metab 1980;51:144 –147. 32. Martin HE, Serebrin R. Effect of adrenal and parathyroid on electrolyte excretion in rat. Horm Metab Res 1970;2:228–232. 33. Ferriss JB, Brown JJ, Cumming AM, Fraser R, Lever AF, Peacock M et al. Primary hyperparathyroidism associated with primary hyperaldosteronism. Acta Endocrinol (Copenh) 1983;103:365 –370. 34. Rodriguez-Ayala E, Avila-Diaz M, Foyo-Niembro E, Amato D, Ramirez-San-Juan E, Paniagua R. Effect of parathyroidectomy on cardiac fibrosis and apoptosis: possible role of aldosterone. Nephron Physiol 2006;103:p112–p118. 35. Pacifici R, Perry HM 3rd, Shieber W, Biglieri E, Droke DM, Avioli LV. Adrenal responses to subtotal parathyroidectomy for primary hyperparathyroidism. Calcif Tissue Int 1987;41:119 –123. 36. Sotornik I, Stribrna J, Hronova J, Kocandrle V, Janata V, Taborsky P et al. Changes in plasma renin and aldosterone after parathyroidectomy in patients with hyperparathyroidism. Cas Lek Cesk 1993;132:45– 49. 37. Kovacs L, Goth MI, Szabolcs I, Dohan O, Ferencz A, Szilagyi G. The effect of surgical treatment on secondary hyperaldosteronism and relative hyperinsulinemia in primary hyperparathyroidism. Eur J Endocrinol 1998;138:543 – 547. 38. Brunaud L, Germain A, Zarnegar R, Rancier M, Alrasheedi S, Caillard C et al. Serum aldosterone is correlated positively to parathyroid hormone (PTH) levels in patients with primary hyperparathyroidism. Surgery 2009;146:1035 –1041. 39. Bernini G, Moretti A, Lonzi S, Bendinelli C, Miccoli P, Salvetti A. Renin-angiotensin-aldosterone system in primary hyperparathyroidism before and after surgery. Metabolism 1999;48:298–300. 40. Jespersen B, Randlov A, Abrahamsen J, Fogh-Andersen N, Kanstrup IL. Effects of PTH(1 –34) on blood pressure, renal function, and hormones in essential hypertension: the altered pattern of reactivity may counteract raised blood pressure. Am J Hypertens 1997;10:1356 –1367. 41. Richards AM, Espiner EA, Nicholls MG, Ikram H, Hamilton EJ, Maslowski AH. Hormone, calcium and blood pressure relationships in primary hyperparathyroidism. J Hypertens 1988;6:747 –752. 42. Salahudeen AK, Thomas TH, Sellars L, Tapster S, Keavey P, Farndon JR et al. Hypertension and renal dysfunction in primary hyperparathyroidism: effect of parathyroidectomy. Clin Sci (Lond) 1989;76:289 –296. 43. Tomaschitz A, Pilz S. Aldosterone to renin ratio—a reliable screening tool for primary aldosteronism? Horm Metab Res 2010;42:382 – 391. 44. Letizia C, Ferrari P, Cotesta D, Caliumi C, Cianci R, Cerci S et al. Ambulatory monitoring of blood pressure (AMBP) in patients with primary hyperparathyroidism. J Hum Hypertens 2005;19:901 –906. 45. Rosa J, Raska I Jr, Wichterle D, Petrak O, Strauch B, Somloova Z et al. Pulse wave velocity in primary hyperparathyroidism and effect of surgical therapy. Hypertens Res 2011;34:296–300. 46. Nilsson IL, Aberg J, Rastad J, Lind L. Endothelial vasodilatory dysfunction in primary hyperparathyroidism is reversed after parathyroidectomy. Surgery 1999;126: 1049–1055. 47. Hulter HN, Melby JC, Peterson JC, Cooke CR. Chronic continuous PTH infusion results in hypertension in normal subjects. J Clin Hypertens 1986;2:360 –370. 48. Schiffl H, Lang SM. Hypertension Secondary to PHPT: Cause or Coincidence?. Int J Endocrinol 2011; doi:10.1155/2011/974647. 49. Brandenburger Y, Kennedy ED, Python CP, Rossier MF, Vallotton MB, Wollheim CB et al. Possible role for mitochondrial calcium in angiotensin II- and potassiumstimulated steroidogenesis in bovine adrenal glomerulosa cells. Endocrinology 1996; 137:5544 –5551. 50. Rossier MF, Burnay MM, Vallotton MB, Capponi AM. Distinct functions of T- and L-type calcium channels during activation of bovine adrenal glomerulosa cells. Endocrinology 1996;137:4817 –4826. 51. Spat A, Hunyady L. Control of aldosterone secretion: a model for convergence in cellular signaling pathways. Physiol Rev 2004;84:489 –539. 52. Matsunaga H, Yamashita N, Maruyama Y, Kojima I, Kurokawa K. Evidence for two distinct voltage-gated calcium channel currents in bovine adrenal glomerulosa cells. Biochem Biophys Res Commun 1987;149:1049 –1054. 18 53. Payet MD, Durroux T, Bilodeau L, Guillon G, Gallo-Payet N. Characterization of K+ and Ca2+ ionic currents in glomerulosa cells from human adrenal glands. Endocrinology 1994;134:2589 –2598. 54. Aritomi S, Wagatsuma H, Numata T, Uriu Y, Nogi Y, Mitsui A et al. Expression of N-type calcium channels in human adrenocortical cells and their contribution to corticosteroid synthesis. Hypertens Res 2011;34:193–201. 55. Wiederkehr A, Szanda G, Akhmedov D, Mataki C, Heizmann CW, Schoonjans K et al. Mitochondrial matrix calcium is an activating signal for hormone secretion. Cell Metab 2011;13:601–611. 56. Cherradi N, Rossier MF, Vallotton MB, Capponi AM. Calcium stimulates intramitochondrial cholesterol transfer in bovine adrenal glomerulosa cells. J Biol Chem 1996; 271:25971 –25975. 57. Massry SG, Fadda GZ. Chronic renal failure is a state of cellular calcium toxicity. Am J Kidney Dis 1993;21:81–86. 58. Startchik I, Morabito D, Lang U, Rossier MF. Control of calcium homeostasis by angiotensin II in adrenal glomerulosa cells through activation of p38 MAPK. J Biol Chem 2002;277:24265 –24273. 59. Barrett PQ, Isales CM, Bollag WB, McCarthy RT. Modulation of Ca2+ channels by atrial natriuretic peptide in the bovine adrenal glomerulosa cell. Can J Physiol Pharmacol 1991;69:1553 –1560. 60. Deicher R, Kirsch B, Mullner M, Kaczirek K, Niederle B, Hörl WH. Impact of parathyroidectomy on neutrophil cytosolic calcium in chronic kidney disease patients: a prospective parallel group trial. J Intern Med 2005;258:67– 76. 61. Miyauchi A, Notoya K, Mikuni-Takagaki Y, Takagi Y, Goto M, Miki Y et al. Parathyroid hormone-activated volume-sensitive calcium influx pathways in mechanically loaded osteocytes. J Biol Chem 2000;275:3335 –3342. 62. Kitazawa S, Fukase M, Kitazawa R, Takenaka A, Gotoh A, Fujita T et al. Immunohistologic evaluation of parathyroid hormone-related protein in human lung cancer and normal tissue with newly developed monoclonal antibody. Cancer 1991;67:984 –989. 63. Urena P, Kong XF, Abou-Samra AB, Juppner H, Kronenberg HM, Potts JT Jr et al. Parathyroid hormone (PTH)/PTH-related peptide receptor messenger ribonucleic acids are widely distributed in rat tissues. Endocrinology 1993;133:617 –623. 64. Klin M, Smogorzewski M, Khilnani H, Michnowska M, Massry SG. Mechanisms of PTH-induced rise in cytosolic calcium in adult rat hepatocytes. Am J Physiol 1994; 267:G754 –G763. 65. Mazzocchi G, Aragona F, Malendowicz LK, Nussdorfer GG. PTH and PTH-related peptide enhance steroid secretion from human adrenocortical cells. Am J Physiol Endocrinol Metab. 2001;280:E209 –E213. 66. Rosenberg J, Pines M, Hurwitz S. Response of adrenal cells to parathyroid hormone stimulation. J Endocrinol 1987;112:431 – 437. 67. Olgaard K, Lewin E, Bro S, Daugaard H, Egfjord M, Pless V. Enhancement of the stimulatory effect of calcium on aldosterone secretion by parathyroid hormone. Miner Electrolyte Metab 1994;20:309–314. 68. Isales CM, Barrett PQ, Brines M, Bollag W, Rasmussen H. Parathyroid hormone modulates angiotensin II-induced aldosterone secretion from the adrenal glomerulosa cell. Endocrinology 1991;129:489 –495. 69. Fallo F, Rocco S, Pagotto U, Zangari M, Luisetto G, Mantero F. Aldosterone and pressor responses to angiotensin II in primary hyperparathyroidism. J Hypertens Suppl 1989;7:S192 –S193. 70. Grant FD, Mandel SJ, Brown EM, Williams GH, Seely EW. Interrelationships between the renin-angiotensin-aldosterone and calcium homeostatic systems. J Clin Endocrinol Metab 1992;75:988 –992. 71. Rafferty B, Zanelli JM, Rosenblatt M, Schulster D. Corticosteroidogenesis and adenosine 3′ , 5′ - monophosphate production by the amino-terminal (1 –34) fragment of human parathyroid hormone in rat adrenocortical cells. Endocrinology 1983;113: 1036 –1042. 72. Tomaschitz A, Pilz S, Ritz E, Grammer T, Drechsler C, Boehm BO et al. Independent association between 1,25-dihydroxyvitamin D, 25-hydroxyvitamin D and the renin-angiotensin system: The Ludwigshafen Risk and Cardiovascular Health (LURIC) study. Clin Chim Acta 2010;411:1354 –1360. 73. Ozata M, Durmus O, Yilmaz MI, Bolu E, Erdogan M, Ozdemir IC. The renin-angiotensin-aldosterone system (RAAS) and its relation with calcium homeostasis in male obesity. Med Sci Monit 2002;8:CR430 –CR434. 74. Mihailidou AS, Loan Le TY, Mardini M, Funder JW. Glucocorticoids activate cardiac mineralocorticoid receptors during experimental myocardial infarction. Hypertension 2009;54:1306 –1312. 75. Yoo YM, Baek MG, Jung EM, Yang H, Choi KC, Yu FH et al. Parathyroid hormone-related protein and glucocorticoid receptor beta are regulated by cortisol in the kidney of male mice. Life Sci 2011;89:615 –620. 76. Dickstein K, Cohen-Solal A, Filippatos G, McMurray JJ, Ponikowski P, Poole-Wilson PA et al. ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2008: the Task Force for the diagnosis and treatment of acute and chronic heart failure 2008 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association of the ESC (HFA) and endorsed by the European Society of Intensive Care Medicine (ESICM). Eur J Heart Fail 2008;10:933 –989. 77. Weber KT. Aldosterone in congestive heart failure. N Engl J Med 2001;345: 1689 –1697. A. Tomaschitz et al. 78. Dell’Italia LJ. Translational success stories: angiotensin receptor 1 antagonists in heart failure. Circ Res 2011;109:437 –452. 79. Tomaschitz A, Pilz S, Ritz E, Meinitzer A, Boehm BO, März W. Plasma aldosterone levels are associated with increased cardiovascular mortality: the Ludwigshafen Risk and Cardiovascular Health (LURIC) study. Eur Heart J 2010;31:1237 –1247. 80. Ritz E, Tomaschitz A. Aldosterone, a vasculotoxic agent—novel functions for an old hormone. Nephrol Dial Transplant 2009;24:2302 –2305. 81. Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999;341:709 –717. 82. Pitt B, Reichek N, Willenbrock R, Zannad F, Phillips RA, Roniker B et al. Effects of eplerenone, enalapril, and eplerenone/enalapril in patients with essential hypertension and left ventricular hypertrophy: the 4E-left ventricular hypertrophy study. Circulation 2003;108:1831 –1838. 83. Zannad F, McMurray JJ, Krum H, van Veldhuisen DJ, Swedberg K, Shi H et al. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364:11–21. 84. Iglarz M, Touyz RM, Viel EC, Amiri F, Schiffrin EL. Involvement of oxidative stress in the profibrotic action of aldosterone. Interaction with the renin-angiotension system. Am J Hypertens 2004;17:597 – 603. 85. Pratt JH. Central role for ENaC in development of hypertension. J Am Soc Nephrol 2005;16:3154 –3159. 86. Runyan AL, Chhokar VS, Sun Y, Bhattacharya SK, Runyan JW, Weber KT. Bone loss in rats with aldosteronism. Am J Med Sci 2005;330:1 –7. 87. Chhokar VS, Sun Y, Bhattacharya SK, Ahokas RA, Myers LK, Xing Z et al. Loss of bone minerals and strength in rats with aldosteronism. Am J Physiol Heart Circ Physiol 2004;287:H2023 –H2026. 88. Law PH, Sun Y, Bhattacharya SK, Chhokar VS, Weber KT. Diuretics and bone loss in rats with aldosteronism. J Am Coll Cardiol 2005;46:142 –146. 89. Chhokar VS, Sun Y, Bhattacharya SK, Ahokas RA, Myers LK, Xing Z et al. Hyperparathyroidism and the calcium paradox of aldosteronism. Circulation 2005;111:871 –878. 90. Kamalov G, Ahokas RA, Zhao W, Zhao T, Shahbaz AU, Johnson PL et al. Uncoupling the coupled calcium and zinc dyshomeostasis in cardiac myocytes and mitochondria seen in aldosteronism. J Cardiovasc Pharmacol 2010;55:248 –254. 91. Massry SG, Smogorzewski M. Mechanisms through which parathyroid hormone mediates its deleterious effects on organ function in uremia. Semin Nephrol 1994; 14:219 –231. 92. Zia AA, Kamalov G, Newman KP, McGee JE, Bhattacharya SK, Ahokas RA et al. From aldosteronism to oxidative stress: the role of excessive intracellular calcium accumulation. Hypertens Res 2010;33:1091 –1101. 93. Shahbaz AU, Kamalov G, Zhao W, Zhao T, Johnson PL, Sun Y et al. Mitochondriatargeted cardioprotection in aldosteronism. J Cardiovasc Pharmacol 2011;57:37 –43. 94. Cheema Y, Sherrod JN, Zhao W, Zhao T, Ahokas RA, Sun Y et al. Mitochondriocentric pathway to cardiomyocyte necrosis in aldosteronism: cardioprotective responses to carvedilol and nebivolol. J Cardiovasc Pharmacol 2011;58:80– 86. 95. Halestrap AP. Calcium, mitochondria and reperfusion injury: a pore way to die. Biochem Soc Trans 2006;34:232–237. 96. Cappuccio FP, Markandu ND, MacGregor GA. Renal handling of calcium and phosphate during mineralocorticoid administration in normal subjects. Nephron 1988;48: 280– 283. 97. Gehr MK, Goldberg M. Hypercalciuria of mineralocorticoid escape: clearance and micropuncture studies in the rat. Am J Physiol 1986;251:F879 –F888. 98. Suki WN. Disposition and regulation of body potassium: an overview. Am J Med Sci 1976;272:31 –41. 99. Lemann J Jr, Piering WF, Lennon EJ. Studies of the acute effects of aldosterone and cortisol on the interrelationship between renal sodium, calcium and magnesium excretion in normal man. Nephron 1970;7:117 –130. 100. Khouzam RN, Dishmon DA, Farah V, Flax SD, Carbone LD, Weber KT. Secondary hyperparathyroidism in patients with untreated and treated congestive heart failure. Am J Med Sci 2006;331:30– 34. 101. Rossi E, Perazzoli F, Negro A, Sani C, Davoli S, Dotti C et al. Acute effects of intravenous sodium chloride load on calcium metabolism and on parathyroid function in patients with primary aldosteronism compared with subjects with essential hypertension. Am J Hypertens 1998;11:8 –13. 102. Benitah JP, Vassort G. Aldosterone upregulates Ca(2+) current in adult rat cardiomyocytes. Circ Res 1999;85:1139 –1145. 103. Vidal A, Sun Y, Bhattacharya SK, Ahokas RA, Gerling IC, Weber KT. Calcium paradox of aldosteronism and the role of the parathyroid glands. Am J Physiol Heart Circ Physiol 2006;290:H286 – H294. 104. Goodwin KD, Ahokas RA, Bhattacharya SK, Sun Y, Gerling IC, Weber KT. Preventing oxidative stress in rats with aldosteronism by calcitriol and dietary calcium and magnesium supplements. Am J Med Sci 2006;332:73–78. 105. Chen J, Normand SL, Wang Y, Krumholz HM. National and regional trends in heart failure hospitalization and mortality rates for Medicare beneficiaries, 1998 –2008. JAMA 2011;306:1669 – 1678. 106. Jessup M, Abraham WT, Casey DE, Feldman AM, Francis GS, Ganiats TG et al. 2009 focused update: ACCF/AHA Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/ Aldosterone and parathyroid hormone 107. 108. 109. 110. 111. 112. 113. 114. 115. 116. 117. 118. 119. 120. 121. American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009;119:1977 –2016. Albert NM, Yancy CW, Liang L, Zhao X, Hernandez AF, Peterson ED et al. Use of aldosterone antagonists in heart failure. JAMA 2009;302:1658 –1665. Maron BA, Leopold JA. Aldosterone receptor antagonists: effective but often forgotten. Circulation 2010;121:934 –939. Funder JW, Carey RM, Fardella C, Gomez-Sanchez CE, Mantero F, Stowasser M et al. Case detection, diagnosis, and treatment of patients with primary aldosteronism: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2008;93: 3266 –3281. Rossi GP, Di Bello V, Ganzaroli C, Sacchetto A, Cesari M, Bertini A et al. Excess aldosterone is associated with alterations of myocardial texture in primary aldosteronism. Hypertension 2002;40:23– 27. Milliez P, Girerd X, Plouin PF, Blacher J, Safar ME, Mourad JJ. Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J Am Coll Cardiol 2005;45:1243 –1248. Sechi LA, Novello M, Lapenna R, Baroselli S, Nadalini E, Colussi GL et al. Long-term renal outcomes in patients with primary aldosteronism. JAMA 2006;295:2638 –2645. Resnick LM, Muller FB, Laragh JH. Calcium-regulating hormones in essential hypertension. Relation to plasma renin activity and sodium metabolism. Ann Intern Med 1986;105:649 –654. Rastegar A, Agus Z, Connor TB, Goldberg M. Renal handling of calcium and phosphate during mineralocorticoid “escape” in man. Kidney Int 1972;2:279–286. Horton R, Biglieri EG. Effect of aldosterone on the metabolism of magnesium. J Clin Endocrinol Metab 1962;22:1187 –1192. Resnick LM, Laragh JH. Calcium metabolism and parathyroid function in primary aldosteronism. Am J Med 1985;78:385 –390. Kabadi UM. Renal calculi in primary hyperaldosteronism. J Postgrad Med 1995;41: 17 –18. Shey J, Cameron MA, Sakhaee K, Moe OW. Recurrent calcium nephrolithiasis associated with primary aldosteronism. Am J Kidney Dis 2004;44:e7 –e12. Bhattacharya SK, Gandhi MS, Kamalov G, Ahokas RA, Sun Y, Gerling IC et al. Myocardial remodeling in low-renin hypertension: molecular pathways to cellular injury in relative aldosteronism. Curr Hypertens Rep 2009;11:412 –420. Rossi E, Sani C, Perazzoli F, Casoli MC, Negro A, Dotti C. Alterations of calcium metabolism and of parathyroid function in primary aldosteronism, and their reversal by spironolactone or by surgical removal of aldosterone-producing adenomas. Am J Hypertens 1995;8:884 –893. Pilz S, Tomaschitz A, Stepan V, Obermayer-Pietsch B, Fahrleitner-Pammer A, Schweighofer N et al. Graz Endocrine Causes of Hypertension (GECOH) study: a diagnostic accuracy study of aldosterone to active renin ratio in screening for primary aldosteronism. BMC Endocr Disord 2009;9:11. 19 122. Pilz S, Kienreich K, Drechsler C, Ritz E, Fahrleitner-Pammer A, Gaksch M et al. Hyperparathyroidism in patients with primary aldosteronism: cross-sectional and interventional data from the GECOH study. J Clin Endocrinol Metab 2012;97:E75–79. 123. Maniero CFA, Seccia TM, Toniato A, Iacobone M, Piebani M, De Caro R et al. Mild hyperparathyroidism: a novel surgically correctable feature of primary aldosteronism. J Hypertens 2012;30:390 –395. 124. Rizk-Rabin MAG, Rene-Corail F, Perlemoine K, Hamzaoui H, Tissier F. Differential expression of parathyroid hormone-related protein in adrenocortical tufmors: autocrine/paracrine effects on the grwoth and signaling pathways in H295R cells. Cancer Epidemiol Biomarkers Prev 2008;17:2275 –2285. 125. Nishioka K, Nishida M, Ariyoshi M, Jian Z, Saiki S, Hirano M et al. Cilostazol suppresses angiotensin II-induced vasoconstriction via protein kinase A-mediated phosphorylation of the transient receptor potential canonical 6 channel. Arterioscler Thromb Vasc Biol 2011;31:2278 –2286. 126. Newfell BG, Iyer LK, Mohammad NN, McGraw AP, Ehsan A, Rosano G et al. Aldosterone regulates vascular gene transcription via oxidative stress-dependent and -independent pathways. Arterioscler Thromb Vasc Biol 2011;31:1871 – 1880. 127. De Giusti VC, Nolly MB, Yeves AM, Caldiz CI, Villa-Abrille MC, Chiappe de Cingolani GE et al. Aldosterone stimulates the cardiac Na(+)/H(+) exchanger via transactivation of the epidermal growth factor receptor. Hypertension 2011;58: 912– 919. 128. Brown MJ. Personalised medicine for hypertension. BMJ 2011;343:d4697. doi: 10.1136/bmj.d4697. 129. Tomaschitz A, Pilz S, Ritz E, Morganti A, Grammer T, Amrein K et al. Associations of plasma renin with 10-year cardiovascular mortality, sudden cardiac death, and death due to heart failure. Eur Heart J 2011;32:2642 –2649. 130. Gaziano TA, Bitton A, Anand S, Weinstein MC. The global cost of nonoptimal blood pressure. J Hypertens 2009;27:1472 –1477. 131. Gomez Sanchez EP. Central mineralocorticoid receptors and cardiovascular disease. Neuroendocrinology 2009;90:245 –250. 132. Blaustein MP, Leenen FH, Chen L, Golovina VA, Hamlyn JM, Pallone TL et al. How NaCl raises blood pressure: a new paradigm for the pathogenesis of salt-dependent hypertension. Am J Physiol Heart Circ Physiol 2011; doi: 10.1152/ajpheart.00899.2011. 133. Hochberg Z. Hormone resistance at the clinical level. Sci Signal 2010;3:pt1. 134. Mulatero P, Tizzani D, Viola A, Bertello C, Monticone S, Mengozzi G et al. Prevalence and characteristics of familial hyperaldosteronism: the PATOGEN study (Primary Aldosteronism in TOrino-GENetic forms). Hypertension 2011;58: 797– 803. 135. Ehret GB, Munroe PB, Rice KM, Bochud M, Johnson AD, Chasman DI et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature 2011;478:103 –109. 136. Fuentes JGP, Modesto T, Rotllant J, Canario AV, Power DM. A PTH/PTHrP receptor antagonist blocks the hyperclacemic response to estradiol-17beta. Am J Physiol Regul Integr Comp Physiol 2007;293:R956– R960.