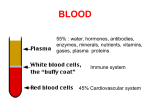
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Page 1 of31 Clotting Factors Clinical Policy Bulletin: Clotting Factors Revised April 2014 Number: 0131 Policy I. Aetna considers anti-hemophilic factor (factor VIII), factor IX (e.g., BeneFIX, and Rixubis), and Humate-P or medically necessary to prevent or treat hemorrhagic complications in adults and children with hemophilia A, B or von Willebrand's disease according to the following criteria and limitations: A. Standard dose therapy (dose until bleeding stops or up to 14 days after surgery) when both of the fol criteria are met: 1. Member has a diagnosis of hemophilia A, hemophilia B, or von Willebrand's disease (only Hu Alphanate may be used in von Willebrand’s disease); and 2. Member is hemorrhaging or physical trauma such as surgery is anticipated (secondary short-t prophylaxis). B. Continuous (long-term) prophylactic therapy when either of the following criteria is met: 1. Primary prophylactic therapy: Member has severe hemophilia A or hemophilia B (less than 1 factor (less than 0.01 IU/ml)); or 2. Secondary prophylactic therapy: Member has hemophilia A or hemophilia B (regardless of no levels) and has documented history of 2 or more episodes of spontaneous bleeding into joints C. High-dose immune tolerance induction (dosing continues beyond 14 days) when all of the following c met: 1. Anti-hemophilic factor or Factor IX survival and recovery of anti-hemophilic factor levels after i abnormal (see D, 1, below); and 2. Attempts to lower antibody levels with either immunosuppressant or corticosteroids have been unsuccessful; and 3. Member has a diagnosis of hemophilia A or hemophilia B; and 4. Member has inhibitors (anti-factor VIII:c or IX:c antibodies). D. Limits applicable to immune tolerance induction: http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014 Page 2 of31 Clotting Factors 1. Continued immune tolerance induction is no longer considered medically necessary when all o following criteria are met: a. Anti-hemophilic factor or factor IX survival after infusion is normal (6-hr level at least 46 min level); and b. Inhibitor levels become undetectable; and c. Recovery of anti-hemophilic factor or factor IX levels after infusion are normal (defined 85 % of the expected for individuals without inhibitors); 2. Cases in which members are on immune tolerance induction for 6 months or more may be ref review of medical necessity to determine whether continued immune tolerance therapy is me necessary. Aetna considers anti-hemophilic factor (factor VIII), factor IX and Humate-P or Alphanate experimental and investigational for all other indications because their effectiveness for indications other than the ones listed a not been established. II. Aetna considers recombinant factor VIII Fc fusion protein (Eloctate) medically necessary to prevent or treat hemorrhagic complications in adults and children with hemophilia A according to the following criteria and li A. Standard dose therapy (dose until bleeding stops or up to 14 days after surgery) when both of the fol criteria are met: 1. Member has a diagnosis of hemophilia A; and 2. Member is hemorrhaging or physical trauma such as surgery is anticipated (secondary short-t prophylaxis). B. Continuous (long-term) prophylactic therapy when either of the following criteria is met: 1. Primary prophylactic therapy: Member has severe hemophilia A (less than 1 % of normal facto 0.01 IU/ml)); or 2. Secondary prophylactic therapy: Member has hemophilia A (regardless of normal factor level documented history of 2 or more episodes of spontaneous bleeding into joints. III. Aetna considers recombinant factor VIIa (rFVIIa, NovoSeven RT) medically necessary for the prevention of surgical interventions or invasive procedures and for the control of bleeding events in members with any of t indications: A. Members with hemophilia A or hemophilia B who have developed inhibitor antibodies to factor VIII or or B. Members with acquired hemophilia or congenital FVII deficiency; or C. Members with Glanzmann's thrombasthenia with antibodies to glycoprotein IIb-IIIa and/or human leu antigen (HLA), and with past or present refractoriness to platelet transfusions. Aetna considers rFVIIa experimental and investigational for all other indications including prevention unrelated to hemophilia or factor deficiency (e.g., in persons undergoing cardiac surgery, liver transp vascular surgery, or prostatectomy); or treatment of bleeding unrelated to hemophilia or factor deficie trauma, acute coronary syndromes, acute spontaneous intracerebral hemorrhage, acute variceal ble gastrointestinal bleeding secondary to Crohn's disease, and intracranial bleeding from traumatic brai http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014 Clotting Factors Page 3 of31 stroke, not an all-inclusive list). IV. Aetna considers human anti-thrombin III (Thrombate III) medically necessary for the treatment of persons w hereditary anti-thrombin III deficiency in connection with thrombo-embolism, obstetrical procedures, or surgi procedures. Aetna considers human anti-thrombin III experimental and investigational for all other indications including it in critically ill individuals. V. Aetna considers factor eight inhibitor bypassing activity (FEIBA) (Febia NF) anti-inhibitor coagulant complex necessary in persons with hemophilia A and B with inhibitors for the control and prevention of bleeding epis perioperative management, and routine prophylaxis to prevent or reduce the frequency of bleeding episodes Aetna considers FEIBA experimental and investigational for all other indications (e.g., reversal of anticoagu associated coagulopathy, rescue treatment of coagulopathy after cardiac surgery; prevention and treatment in non-hemophilic persons with acquired inhibitors, not an all-inclusive list) because its effectiveness for indi other than the ones listed above has not been established. VI. Aetna considers factor XIII concentrate [Human] (Corifact) medically necessary for the prevention of bleedin the perioperative management of surgical bleeding in persons with congenital factor XIII deficiency. Aetna considered factor XIII concentrate experimental and investigational for all other indications because it effectiveness for indications other than the one listed above has not been established. VII. Aetna considers recombinant coagulation factor XIII A-subunit (Tretten) medically necessary for routine prop bleeding in persons with congenital factor XIII A-subunit deficiency. Coagulation factor XIII A-subunit is considered experimental and investigational for persons with congenital -subunit deficiency and for all other indications. VIII. Aetna considers von Willebrand factor/coagulation factor VIII complex (Wilate®) medically necessary for the of spontaneous and/or trauma-induced bleeding episodes in individuals with severe von Willebrand disease individuals with mild or moderate VWD when there is failure, contraindication or intolerance to desmopressi Aetna considers von Willebrand factor/coagulation factor VIII complex (Wilate®) experimental and investiga individuals with hemophilia A; and for the prophylaxis of spontaneous bleeding episodes, or the prevention o bleeding during and after surgery in individuals with VWD. IX. Aetna considers prothrombin complex concentrate (human) (Kcentra) medically necessary for urgent revers acquired coagulation factor deficiency induced by vitamin K antagonist (e.g., warfarin) therapy in adults with major bleeding. Aetna considers prothrombin complex concentrate (human) experimental and investigational for reversal of coagulation in persons without acute major bleeding; and trauma-induced hemorrhage because its effective these indications has not been established. X. Aetna considers recombinant factor IX fc fusion protein (rFIXFc) (Alprolix) medically necessary for persons d with hemophilia B (endogenous factor IX level of ≤2 IU per deciliter, or ≤2% of normal levels) for the followin indications: 1) control and prevention of bleeding episodes, 2) perioperative management, and 3) routine pro prevent or reduce the frequency of bleeding episodes. A. Standard dose therapy (dose until bleeding stops or up to 14 days after surgery) when both of the fol criteria are met: http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014 Page 4 of31 Clotting Factors 1. Member has a diagnosis of hemophilia B; and 2. Member is hemorrhaging or physical trauma such as surgery is anticipated (secondary short-t prophylaxis). B. Continuous (long-term) prophylactic therapy when either of the following criteria is met: 1. Primary prophylactic therapy: Member has severe hemophilia B (less than 1 % of normal facto 0.01 IU/ml)); or 2. Secondary prophylactic therapy: Member has hemophilia B (regardless of normal factor levels documented history of 2 or more episodes of spontaneous bleeding into joints. Aetna considers rFIXFc experimental and investigational for induction of immune tolerance in with hemophilia B and for all other indications. Note: This policy applies only to clotting factor products available through pharmaceutical suppliers. Cryoprecipitat factors are available only through blood banks and have slightly different indications. Background Hemophilia and von Willebrand's disease are the most common congenital bleeding disorders. Hemophilia refers bleeding disorders in which there is a deficiency (activity level of 35 % or less) of either factor VIII (hemophilia A, cl hemophilia) or factor IX (hemophilia B, Christmas disease). In general, administration of anti-hemophilic factor is in hemophilia when a bleeding episode arises (demand treatment) or when bleeding is anticipated or likely (prophyla treatment). Primary prophylactic therapy may be indicated for patients with severe hemophilia A or hemophilia B who have les of normal factor (less than 0.01 IU/ml; National Hemophilia Foundation, 2001). Primary prophylactic therapy shoul instituted early, prior to the onset of frequent bleeding, with the aim of keeping the trough factor or VIII or factor IX l 1 % between doses (National Hemophilia Foundation, 2001). In some cases, continuous prophylactic therapy ma indicated in persons with hemophilia A or hemophilia B that is not severe (i.e., hemophiliacs with more than 1 % of factor levels) who have repeated episodes of spontaneous bleeding. Short-term prophylactic treatment is given to patients before they undergo surgical procedures or engage in activiti carry a high risk of provoking a bleed. It may also be given to break the cycle of frequent bleeding into specific join joints). Potential harm, including the risk of hepatitis B, hepatitis C and HIV infection, has now been minimized thro inactivation of plasmaderived coagulation-factor concentrates and through the use of recombinant clotting factor c and other non-plasma-derived hemostatic agents. Currently, cryoprecipitate does not undergo viral inactivation pro Immune tolerance induction is designed to overcome the effects of anti-hemophilic factor or factor IX inhibitors in c hemophiliac patients, thus restoring effectiveness of antihemophilic factor or factor IX therapy to resolve active ble these patients. It consists of administration of very high doses of anti-hemophilic factor or factor IX over an extend time. A number of different immune tolerance therapy regimens have been developed using replacement factor VIII or fa produce long-term inhibitor suppression or eradication. These protocols utilize various doses of replacement facto factor IX with or without additional therapies. http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014 Clotting Factors Page 5 of31 There are a number of established protocols for immune tolerance induction; these regimens differ in dose and dur therapy (Stachnik, 2003). The North American Immune Tolerance Registry was established in 1993 to assess the protocols used for immune tolerance therapy in the United States and Canada (Stachnik, 2003; Kroner, 1999; DiM Kroner, 1999). Based on a survey of 168 centers that provide medical care to hemophilia patients, many clinicians America (about 50 %) use 100 to 200 U/kg of replacement factor VIII daily for immune tolerance therapy. In gener duration of treatment is shorter when using higher doses of factor VIII. For doses of factor VIII over 50 U/kg daily, treatment time was between 6 and 7 months; however, with lower doses of factor VIII (less than 50 U/kg daily), the duration of treatment was nearly 19 months. Highest success rates were found among patients who had a history maximum inhibitor titers (less than 50 Bethesda Units). Immune tolerance induction is not successful in all patients -- approximately 20 % to 30 % fail to respond (Stachnik Although the duration of treatment varies among the immune tolerance protocols, most clinicians would agree that who has not responded as expected after 12 to 24 months of treatment is unlikely to respond with further treatmen induction tolerance, although occasionally successful, generally is not effective (Stachnik, 2003). For these patient episodes can usually be controlled using bypassing agents, including factor IX complex (prothrombin complex con PCCs), recombinant factor VIIa (rFVIIa, NovoSeven RT), or activated anti-inhibitor coagulant complexes (AICCs or In view of the increasing safety of clotting-factor concentrates, long-term prophylactic therapy in the form of factor at least 3 times a week or factor IX infusion at least twice a week to prevent hemarthrosis in severely affected patie gaining acceptance, especially in the treatment of infants and children. It has been shown that increasing in-vivo c factor levels to more than 1 % activity (usually accomplished by giving 25 to 40 U/kg of factor VIII 3 times a week o U/kg of factor IX twice a week) is sufficient to prevent most spontaneous joint bleeds and preserve joint function. von Willebrand's disease is typically mild and it generally exhibits an autosomal dominant pattern of inheritance. A hemophilic factor/von Willebrand factor complex (Humate-P) is indicated for use in adult patients for treatment and of bleeding in hemophilia A and in adult and pediatric patients for treatment of spontaneous and trauma-induced bl episodes in severe von Willebrand disease and in mild and moderate von Willebrand disease where use of desmo known or suspected to be inadequate. In April 2007, the U.S. Food and Drug Administration (FDA) approved Hum the prevention of excessive bleeding during and after surgery in certain patients with mild-to-moderate and severe Willebrand disease. Recombinant antihemophilic factor -- plasma/albumin free method (rAHF-PFM) (Advate, Baxter Healthcare, Westl CA) is produced using a protein-free manufacturing process. Based on clinical studies submitted to the FDA comp PFM to a standard recombinant antihemophilic factor (rAHF) (Recombinate, Baxter Healthcare), the FDA has conc rAHF-PFM is therapeutically equivalent to rAHF. Human or animal plasma proteins or albumin are used in the cell process of rAHF, with the theoretical risk of transmission viruses, prions or other pathogens (Schesinger and Ragn Adis, 2003). The Medical and Scientific Advisory Council (MASAC) of the National Hemophilia Foundation has rec that "all efforts should be made to remove human albumin from recombinant factor VIII products" and that "increase should be made to eliminate human and bovine proteins from the manufacturing process of recombinant products. there are no documented episodes of transmission of HIV, West Nile Virus, Creutzfeld-Jacob Disease or other suc pathogens from rAHF products currently on the market. Case reports have been published on the successful use of rFVIIa in patients with Glanzmann's thrombasthenia, w inherited hemorrhagic disorder characterized by a severe reduction in, or absence of, platelet aggregation in respo multiple physiologic agonists due to qualitative or quantitative abnormalities of platelet glycoprotein IIb-IIIa. The E Medicine’s Agency (EMEA) has approved rFVIIa for use in persons with Glanzmann’s thrombasthenia with antibod glycoprotein IIb-IIIa and/or human leukocyte antigen (HLA), and with past or present refractoriness to platelet trans scientific discussion prepared by the EMEA (2005) states that the clinical experience with the treatment of patients Glanzmann’s thrombasthenia with rFVIIa is based on data from a clinical trial (n = 4), an International Registry on Recombinant Factor VIIa and Congenital Platelet Disorders (n = 59), and 10 published case reports. Most of the re clinical experience with the use of rFVIIa in persons with Glanzmann’s thrombasthenia is in patients without anti-gl http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014 Clotting Factors Page 6 of31 antibodies and platelet refractoriness. The EMEA analyzed the efficacy data for persons with anti-glycoprotein IIb/ anti-HLA antibodies and platelet refractoriness. Seventeen patients with from the International Registry and 2 patie independently published case reports were identified, with a total of 40 evaluable bleeding episodes. The EMEA s discussion reports that rFVIIa was effective for bleeding prophylaxis in 18 (95 %) of 19 evaluable (minor plus major procedures. The EMEA states that rFVIIa was effective in stopping 28/40 (70 %) of the evaluable bleeding episode similar as the efficacy of rFVIIa in bleeding episodes reported in Glanzmann’s thrombasthenia patients generally. scientific discussion states that 3 recurrent bleedings (3/40, 8 %) could be successfully treated with additional dose (but in 1 case a concomitant platelet transfusion was given). There is a lack of reliable evidence of the effectiveness of rFVIIa for non-hemophilic indications. Levi and colleagu performed a systematic review on the safety and effectiveness of rFVIIa in patients with or without coagulation dis They concluded that more randomized controlled clinical trials are needed to evaluate the safety and effectiveness for patients without a preexistent coagulation disorder and with severe bleeding. In the meantime, off-labeled use may be considered in patients with life-threatening bleeding. This is in agreement with the observations of Lam et who stated that until further prospective controlled data are available, it is recommended that conventional interven prevention and control of hemorrhage in nonhemophiliac patients should remain the standard of care. More recen O’Connell and colleagues (2006) reported that the use of rFVIIa is associated with severe adcerse events (AEs) e when it is used for off-labeled indications. They analyzed 431 incidents of AEs reported to the FDA during the first after the approval of NovoSeven. They noted that most reported thromboembolic AEs followed the use of the drug labeled indications (n = 151) and occurred in the arterial and venous systems, often resulting in serious morbidity a mortality. The majority of complications occurred within 24 hours of taking the drug. These investigators conclude randomized controlled studies are needed to ascertain the safety and effectiveness of rFVIIa in patients without he A number of studies have found that hemostatic therapy with rFVIIa may reduce growth of hematoma but does not survival or functional outcome after intra-cerebral hemorrhage. In a prospective, randomized, placebo-controlled, d escalation study, Narayan and colleagues (2008) evaluated the safety and preliminary effectiveness of rFVIIa to li intra-cerebral hemorrhage (tICH) progression. Patients were enrolled if they had tICH lesions of at least 2 ml on a computed tomographic scan obtained within 6 hours of injury. Recombinant factor VIIa or placebo was administere hours of the baseline computed tomographic scan but no later than 7 hours after injury. Computed tomographic sc repeated at 24 and 72 hours. Five escalating dose tiers were evaluated (40, 80, 120, 160, and 200 microg/kg rFVI evaluations and AEs were recorded until day 15. No significant differences were detected in mortality rate or numb of AEs among treatment groups. Asymptomatic deep vein thrombosis, detected on routinely performed ultrasound was observed more frequently in the combined rFVIIa treatment group (placebo, 3 %; rFVIIa, 8 %; not significant). significant trend for rFVIIa dose-response to limit tICH volume increase was observed (placebo, 21.0 ml; rFVIIa, 10 authors concluded that in this first prospective study of rFVIIa in tICH, there appeared to be a trend toward less he progression in rFVIIa-treated patients (80 to 200 microg/kg) compared with that seen in placebo-treated patients. T stated that the potential significance of this biological effect on clinical outcomes and the significance of the somew incidence of ultrasound-detected deep vein thromboses in the rFVIIa-treated group need to be examined in a large prospective randomized clinical trial. Mayer et al (2008) carried out a phase 3 trial to evaluate whether rFVIIa reduces growth of hematoma and improve and functional outcomes in patients with ICH. A total of 841 patients were randomly assigned to receive placebo (n microg of rFVIIa per kilogram of body weight (n = 276), or 80 microg of rFVIIa per kilogram (n = 297) within 4 hours onset of stroke. The primary end point was poor outcome, defined as severe disability or death according to the m Rankin scale 90 days after the stroke. Treatment with 80 microg of rFVIIa per kilogram resulted in a significant red growth in volume of the hemorrhage. The mean estimated increase in volume of the ICH at 24 hours was 26 % in group, as compared with 18 % in the group receiving 20 microg of rFVIIa per kilogram (p = 0.09) and 11 % in the g receiving 80 microg (p < 0.001). The growth in volume of ICH was reduced by 2.6 ml (95 % confidence interval [C 5.5; p = 0.08) in the group receiving 20 microg of rFVIIa per kilogram and by 3.8 ml (95 % CI: 0.9 to 6.7; p = 0.009) group receiving 80 microg, as compared with the placebo group. Despite this reduction in bleeding, there was no s difference among the 3 groups in the proportion of patients with poor clinical outcome (24 % in the placebo group, http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014 Clotting Factors Page 7 of31 group receiving 20 microg of rFVIIa per kilogram, and 29 % in the group receiving 80 microg). The overall frequen thromboembolic serious AEs was similar in the 3 groups; however, arterial events were more frequent in the group 80 microg of rFVIIa than in the placebo group (9 % versus 4 %, p = 0.04). The authors concluded that hemostatic rFVIIa reduced growth of the hematoma but did not improve survival or functional outcome after ICH. Whether this effect can translate to clinical benefit in a subgroup of patients at high risk for active bleeding, either by treatment w earlier time window or by demonstration of intra-hematomal contrast extravasation after CT angiography deserves study. A randomized controlled study found no effect of rFVIIa on a primary composite endpoint of failure to control 24-hr bleeding, failure to control rebleeding or death in patients with advanced cirrhosis. In a randomized controlled stud and associates (2008) evaluated the safety and effectiveness of rFVIIa in patients with advanced cirrhosis and acti bleeding. At 31 hospitals in an emergency setting, 256 patients (Child-Pugh greater than 8; Child-Pugh B = 26 %, were randomized equally to: placebo; 600 microg/kg rFVIIa (200 + 4x 100 microg/kg); or 300 microg/kg rFVIIa (200 microg/kg). Dosing was intravenous at 0, 2, 8, 14, and 20 hrs after endoscopy, in addition to standard vasoactive, antibiotic, and endoscopic treatment. The primary composite endpoint consisted of failure to control 24-hr bleedin to prevent re-bleeding or death at day 5. Secondary endpoints included AEs and 42-day mortality. Baseline chara were comparable between groups. Administration of rFVIIa had no significant effect on the composite endpoint co placebo (p = 0.37). There was no significant difference in 5-day mortality between groups; however, 42-day morta significantly lower with 600 microg/kg rFVIIa compared with placebo (odds ratio 0.31, 95 % CI: 0.13 to 0.74), and b related deaths were reduced from 12 % (placebo) to 2 % (600 microg/kg). A marked heterogeneity in the failure ra treatment groups was observed across participating centers. Adverse events, including overall thromboembolic ev comparable between groups. The authors concluded that treatment with rFVIIa had no significant effect on the pri composite endpoint compared with placebo. Thus, decision on the use of rFVIIa in acute variceal bleeding should considered, because results of this study do not support the routine use of rFVIIa in this setting. A Cochrane systematic evidence review (Marti-Carvajal et al, 2007) concluded that they found no evidence that rFV reduces the risk of death in patients with liver disease and upper gastrointestinal bleeding. "More randomised clini having low risk of bias are necessary in order to determine the role of human recombinant factor VIIa in clinical pra An assessment by the Canadian Coordinating Office for Health Technology Assessment (Selin and Tejani, 2006) r rFVIIa is increasingly used to control bleeding in non-hemophilic patients during surgery, or during treatment for se or ICH. The assessment noted that there is potential for non-hemophilic use of rFVIIa, particularly if clinical and co effectiveness are established. The assessment concluded that adequately powered randomized controlled trials a to clarify the efficacy and safety of rFVIIa for non-bleeding disorder indications. The assessment noted that phase ICH and trauma are underway. A randomized controlled clinical study found that propylactic use of rFVIIa did not decrease perioperative blood los with normal hemostasis undergoing surgical repair of traumatic pelvic-acetabular fracture. In a double-blind, rando placebocontrolled trial, Raobaikady and associates (2005) examined the clinical value of rFVIIa in patients underg reconstruction surgery for traumatic fracture of pelvis or pelvis and acetabulum (n = 48). Patients were randomized an intravenous bolus injection of rFVIIa 90 microg/kg or placebo as add-on therapy at the time of the first skin incis patients also received intraoperative salvaged red blood cells (RBC). There was no significant difference in the to of peri-operative blood loss, the primary outcome variable, between the rFVIIa and placebo groups. In addition, th differences between the 2 groups in the total volume of blood components, including salvaged RBC transfused, nu patients requiring allogeneic blood components, total volume of fluids infused, total operating time, time taken after intensive care unit to reach normal body temperature and acid-base status, and time spent in hospital. No advers particular thromboembolic events, were reported in either group. The authors concluded that in patients with norm hemostasis undergoing repair surgery of traumatic pelvic-acetabular fracture, the prophylactic use of rFVIIa does n the volume of peri-operative blood loss. A review found that the evidence does not support the use of rFVIIa to prevent or control excessive bleeding after surgery. Hardy and co-workers (2009) noted that excessive bleeding is a common and morbid problem after cardia http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014 Clotting Factors Page 8 of31 There is no doubt a need for an effective and safe hemostatic agent in order to minimize transfusions and avoid su intervention for hemostasis. Recombinant activated factor VII is being used (off-label) increasingly after cardiac su prevent or to control hemorrhage, but its efficacy and safety remain unclear. Several case reports, case series and would tend to support the use of rFVIIa to control excessive bleeding after cardiac operations. On the contrary, 2 r controlled trials have produced negative results whereas a 3rd has not been published yet. Adverse thrombotic ev been reported with increasing frequency. The authors concluded that at present, the generalized use of rFVIIa to control excessive bleeding after cardiac surgery can not be recommended. Analaysis of a large cohort of persons in placebo-controlled trials of rFVIIa found that treatment with high doses of an offlabel basis significantly increased the risk of arterial thromboembolic events. Levi and colleagues (2010) eva rate of thromboembolic events in all published randomized, placebo-controlled trials of rFVIIa used on an off-label non-hemophilia). These investigators analyzed data from 35 randomized clinical trials (26 studies involving patien studies involving healthy volunteers) to determine the frequency of thromboembolic events. The data were pooled use of random-effects models to calculate the odds ratios and 95 % CI. Among 4,468 subjects (4,119 patients and healthy volunteers), 498 had thromboembolic events (11.1 %). Rates of arterial thromboembolic events among all subjects were higher among those who received rFVIIa than among those who received placebo (5.5 % versus 3.2 0.003). Rates of venous thromboembolic events were similar among subjects who received rFVIIa and those who placebo (5.3 % versus 5.7 %). Among subjects who received rFVIIa, 2.9 % had coronary arterial thromboembolic e compared with 1.1 % of those who received placebo (p = 0.002). Rates of arterial thromboembolic events were hi subjects who received rFVIIa than among subjects who received placebo, particularly among those who were 65 y or older (9.0 % versus 3.8 %, p = 0.003); the rates were especially high among subjects 75 years of age or older (1 versus 4.1 %, p = 0.02). The authors concluded that in a large and comprehensive cohort of persons in placebo-c trials of rFVIIa, treatment with high doses of rFVIIa on an off-label basis significantly increased the risk of arterial bu venous thromboembolic events, especially among the elderly. In an editorial that accompanied the afore-mentioned study, Aledort (2010) stated that "[t]his article uniquely prese the offlabel use of rFVIIa in a variety of clinical conditions, and it specifically addresses the incidence of thromboem events. In patients with bleeding disorders, the challenge is to establish hemostasis without incurring thromboemb Clinical trials are put in place to evaluate the safety and efficacy of new agents. Data and safety monitoring boards independent entities, are charged with the responsibility of determining the safety of drugs in a given clinical trial. I determines that the data demonstrate that the risk outweighs the benefit, the trial may be halted, but the reasons fo the trial may not be widely promulgated. Even if trials are not discontinued, regulators, such as the Food and Drug Administration or the European Medicines Agency, when reviewing a portfolio of information about a drug, may not label indication because of adverse events; however, the data on the adverse events often do not reach the public Therefore, patients and health care professionals may have no way to learn about adverse events associated with use". Furthermore, the editorialist stated that "[t]he authors appropriately warn readers that these data warrant scrutiny w is used on an off-label basis. The thrombotic sequelae reported here are not inconsequential. The risk is particula among older patients. This article should serve as a template for pharmaceutical companies to report all studies in use of a given drug, onlabel and off-label, so that physicians can fully appreciate the benefit and risks when makin therapeutic decisions". The Canadian Agency for Drugs and Technologies in Health's technology assessment on rFVIIa for the prevention unrelated to hemophilia (Murphy et al, 2010) concluded that "no consistent benefit of rFVIIa therapy was detected a studies evaluating the prevention of bleeding in patients undergoing prostatectomy, liver transplantation, or cardiac The risk of adverse events after the prophylactic use of rFVIIa in surgical patients is unknown. No conclusions can on the effectiveness or safety of using rFVIIa in the prevention of bleeding in patients who have received supra-the doses of anticoagulant agents. When used for prevention of bleeding, no specific method is available to monitor th effectiveness of rFVIIa". http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014 Clotting Factors Page 9 of31 Mannucci et al (2010) stated that the main cause of the hemostasis defects and related bleeding complications in p acute coronary syndromes (ACS) are the intake of multiple anti-thrombotic drugs, alone or concomitantly with inva procedures such as coronary angiography and percutaneous coronary intervention (PCI). Anti-thrombotic drugs th several phases of hemostasis (platelet function, coagulation, and fibrinolysis) are causing bleeding particularly in e patients, in those who are under-weight and with co-morbidities such as renal insufficiency, diabetes, hypertension malignancy. Identification of patients at high risk of bleeding is the most important preventive strategy, because th and dosages of drugs may to some extent be tailored to the degree of risk. Transfusions of blood products, which become necessary in patients with major bleeding, should be used with caution, because they are associated with cardiovascular events. To reduce the need of transfusion, the hemostatic drugs that decrease blood loss and trans requirements in cardiac surgery (anti-fibrinolytic amino acids, desmopressin, and rFVIIa) might be considered. Ho efficacy of these drugs in the control of bleeding complications is not unequivocally established in ACS and there is for an increased risk of thrombosis. The authors concluded that evidencebased recommendations for the manag bleeding in patients with ACS are currently lacking, so that prevention through accurate assessment of the individu most valid strategy. A Cochrane review found no reliable evidence from randomized controlled clincial trials to support the effectivenes hemostatic drugs in reducing mortality or disability in patients with traumatic brain injury. Perel et al (2010) evaluat effects of hemostatic drugs on mortality, disability and thrombotic complications in patients with traumatic brain inju These investigators included published and unpublished randomized controlled trials comparing hemostatic drugs fibrinolytics: aprotinin, tranexamic acid (TXA), aminocaproic acid or rFVIIa) with placebo, no treatment, or other tre patients with acute TBI. Two review authors independently examined all electronic records, and extracted the data judged that there was clinical heterogeneity between trials so they did not attempt to pool the results of the included results are reported separately. Two trials were included; one was a post-hoc analysis of 30 TBI patients from a ra controlled trial of rFVIIa in blunt trauma patients. The risk ratio for mortality at 30 days was 0.64 (95 % CI: 0.25 to 1 rFVIIa compared to placebo. This result should be considered with caution as the subgroup analysis was not pre- the trial. The other trial evaluated the effect of rFVIIa in 97 TBI patients with evidence of intra-cerebral bleeding in tomography (CT) scan. The corresponding risk ratio for mortality at the last follow-up was 1.08 (95 % CI: 0.44 to 2 quality of the reporting of both trials was poor so it was difficult to assess the risk of bias. The authors concluded th no reliable evidence from randomized controlled trials to support the effectiveness of hemostatic drugs in reducing disability in patients with TBI. New randomized controlled trials assessing the effects of hemostatic drugs in TBI p should be conducted. These trials should be large enough to detect clinically plausible treatment effects. Human AT III (Thrombate III) is indicated for the treatment of patients with hereditary AT III deficiency in connection surgical or obstetrical procedures, or when they suffer from thromboembolism. Rogers (2009) stated that anti-thrombin (AT) functions as a potent natural anticoagulant and serine protease inhibi inactivates many enzymes in the coagulation cascade. Antithrombin also possesses anti-inflammatory properties, which are mediated by its actions as an anti-coagulant. Hereditary AT deficiency is a rare, under-recognised medi that is associated with inadequate endogenous anti-coagulation thought to result from impaired inhibition of serine coagulation factors. Inherited as an autosomal dominant trait, congenital AT deficiency typically reduces functiona to 40 to 60 % of normal. As a result, individuals with hereditary AT deficiency have a greater than or equal to 50 % of venous thromboembolism (VTE). Specifically, AT deficiency is associated with a 3- to 7-fold higher risk of VTE with other thrombophilias. Thus, maintaining adequate levels of AT during high-risk periods is an important treatme Long-term anti-coagulant thrombo-prophylaxis is not recommended in asymptomatic patients with AT deficiency b the increased risk of hemorrhage. However, treatment guidelines recommend short-term thromboprophylaxis in hi clinical settings, including surgery, trauma, and management of pregnancy, labor, and delivery. The goal of treatm patients with hereditary AT deficiency is an initial increase in AT activity to greater than or equal to 120 % of norma followed by maintenance of AT activity at greater than or equal to 80 % of normal levels. Plasma-derived AT, hepa frozen plasma, and human recombinant AT are treatment options for individuals with hereditary AT deficiency. http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014 Clotting Factors Page 10 of31 Tiede et al (2008) stated that during surgery and childbirth, patients with hereditary AT deficiency are at high risk fo thrombosis, and heparin prophylaxis may not be sufficiently effective. In these patients, exogenous AT may be use association with heparin. A recombinant human AT has been developed. In a phase III multi-center study, these i assessed the safety and effectiveness of prophylactic intravenous administration of AT alfa to hereditary AT deficie in high risk situations, including elective surgery, childbirth, or cesarean section. Anti-thrombin alfa was administer and during the high risk period for restoration and maintenance of AT activity at 100 % of normal. Heparin, low-mo weight heparin, and/or vitamin K antagonists were used according to standard of care. The primary efficacy endpo incidence of acute deep vein thrombosis (DVT) from baseline up to day 30 post dosing as assessed by independe review of duplex ultrasonograms and/or venograms. Safety was assessed based on AEs and laboratory evaluatio surgical and 9 obstetrical hereditary AT deficiency patients received AT alfa for a mean period of 7 days. No clinica DVT occurred. Central review of ultrasonograms identified signs of acute DVT in 2 out of 13 evaluable patients. N related AEs were reported. No patient developed anti-AT alfa antibodies. The authors concluded that these finding suggested that AT alfa is a safe and effective alternative to human plasma-derived AT for treating hereditary AT de patients at high risk for thromboembolic events. A Cochrane review concluded that AT III can not be recommended for critically ill patients based on the available e Afshari et al (2008) noted that AT III is an anti-coagulant with anti-inflammatory properties but the efficacy and any effects of AT III supplementation in critically ill patients are unknown. These researchers evaluated the benefits an AT III in critically ill patients. They searched the Cochrane Central Register of Controlled Trials (CENTRAL) (The C Library); MEDLINE; EMBASE; Science Citation Index Expanded; International Web of Science; CINAHL; LILACS; Chinese Biomedical Literature Database (up to November 2006); and contacted authors and manufacturers in the included all randomized clinical trials, irrespective of blinding or language, that compared AT III with no interventio in critically ill patients. The primary outcome measure was mortality. These investigators each independently abst and resolved any disagreements by discussion. They presented pooled estimates of the intervention effects on di outcomes as relative risks (RR) with 95 % CI; performed subgroup analyses to assess risk of bias, the effect of AT different populations (sepsis, trauma, obstetric, and pediatric patients), and the effect of AT III in patients with or wi use of concomitant heparin. They assessed the adequacy of the available number of participants and performed a sequential analysis to establish the implications for further research. These researchers included 20 randomized t total of 3,458 participants; 13 of these trials had high risk of bias. When they combined all trials, AT III did not stati significantly reduce overall mortality compared with the control group (RR 0.96, 95 % CI: 0.89 to 1.03; no heteroge between trials). A total of 32 subgroup and sensitivity analyses were carried out. Analyses based on risk of bias, d populations, and the role of adjuvant heparin gave insignificant differences. Anti-thrombin III reduced the multi-org score among survivors in an analysis involving very few patients. It increased bleeding events (RR 1.52, 95 % CI: 1.78). The authors concluded that AT III can not be recommended for critically ill patients based on the available e The authors stated that a randomized controlled trial of AT III, without adjuvant heparin, with prespecified inclusion good bias protection is needed. Anti-inhibitor coagulant complex, factor eight inhibitor bypassing activity-vapor heated (FEIBA VH), is indicated for of spontaneous bleeding episodes or to cover surgical interventions in hemophilia A and hemophilia B patients with In addition, the use of FEIBA VH has been described in a few non-hemophiliacs with acquired inhibitors to factors XII. The development of inhibitory antibodies to factor VIII is a serious complication of hemophilia. Two hemostatic age different bypassing mechanisms have been used in the treatment of patients with inhibitors: (i) rFVIIa and (ii) activ prothrombin complex concentrate (APCC). Berntorp (2009) noted that the bypassing agents FEIBA anti-inhibitor c complex and rFVIIa have been established as safe and effective therapies for treating bleeding episodes in hemop patients with inhibitors. However, the efficacy of each bypassing agent can vary, and neither agent is universally e The reasons for such variability have yet to be confirmed, but may involve patient-specific factors and the mechani action and pharmacokinetic profiles of these 2 agents. Gomperts et al (2008) noted that FEIBA VH and rFVIIa are available to circumvent the need for factor FVIII in hemophilia A patients with inhibitors, yet their hemostatic efficac http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014 Clotting Factors Page 11 of31 unpredictable. As the results of the FEIBA NovoSeven study illustrated, patients may respond better to one bypas than the other. Furthermore, guidelines from an expert panel reflect that responsiveness to bypassing therapy may from one bleed to the next in the same patient and even from hour to hour during the course of a single bleeding e findings underscored the need to have both bypassing products available to treat bleeding episodes in inhibitor pat frequently evaluate the efficacy of hemostasis during the course of a bleeding event, and to switch products early i response to treatment is unsatisfactory. A cost-effectiveness analysis found that a rFVIIa regimen appears to be a less expensive treatment option in inhibi with minorto-moderate bleeds. Joshi et al (2006) compared the cost-effectiveness of 3 treatment regimens using APCC (FEIBA vapor heated), for home treatment of minor-to-moderate bleeds in hemophilia patients with inhibitor regimens consisting of 1st-, 2nd-, and 3rd-line treatments were: rFVIIa-rFVIIa-rFVIIa; APCC-rFVIIa-rFVIIa; and APC rFVIIa. Patients not responding to 1stline treatment were administered 2nd-line treatment, and those failing 2nd-li 3rd-line treatment. Using literature and expert opinion, the model structure and base-case inputs were adapted to from a previously published analysis. The percentage of evaluable bleeds controlled with rFVIIa and APCC were from published literature. Drug costs (2005 US$) based on average wholesale price were included in the base-cas Uni-variate and probabilistic sensitivity analyses (2nd-order Monte Carlo simulation) were conducted by varying th -bleeding rates, patient weight, and dosing to ascertain robustness of the model. In the base-case analysis, the av per resolved bleed using rFVIIa as 1st-, 2nd-, and 3rd-line treatment was $28,076. Using APCC as 1st-line and rF and 3rd-line treatment resulted in an average cost per resolved bleed of $30,883, whereas the regimen using APC and 2nd-line, and rFVIIa as 3rd-line treatment was the most expensive, with an average cost per resolved bleed of Cost offsets occurred for the rFVIIaonly regimen through avoidance of 2nd and 3rd lines of treatment. In probabili sensitivity analyses, the rFVIIa-only strategy was the least expensive strategy more than 68 % of the time. The au concluded that the management of minor-to-moderate bleeds extends beyond the initial line of treatment, and shou the economic impact of re-bleeding and failures over multiple lines of treatment. In the majority of cases, the rFVII regimen appears to be a less expensive treatment option in inhibitor patients with minor-to-moderate bleeds over 3 treatment. Steen Carlsson et al (2008) compare cost and outcome of APCC and rFVIIa in the treatment of joint bleeds. The a were based on the FENOC (FEIBA NovoSeven Comparative Study) cross-over study where 48 patients used APC rFVIIa to treat 2 joint bleeds. Incremental cost-effectiveness ratios were calculated for 3 outcome measures and th in cost was analyzed using 2 alternative regression methods. Results were subjected to sensitivity analyses. Key determinants of cost were prescribed dose, body weight and treatment in addition to protocol. The cost of APCC w average lower than rFVIIa. At all but one timepoint, patients rated slightly higher (but not statistically significantly) percentages of treatment efficacy and stopping of the bleed by APCC. The reported reduction in pain from start of up to 48 hours varied considerably among individuals. The different relative prices in the U.S., Turkey and Sweden but did not reverse the main results. The authors concluded that the cost per episode was significantly lower for A large individual-level variation in reduction of pain supports decisions that consider the individual patient's experien accept trade-offs between cost and reduction in pain rather than focusing on cost only. In an unified Bayesian meta-regression model, Treur et al (2009) analyzed the published efficacy of rFVIIa and/or A -demand treatments for joint bleeds in hemophilia patients with inhibitors. A systematic search was carried out to i studies reporting on dosage and efficacy of rFVIIa and APCC in the treatment of joint bleeds in the target patient p Data were abstracted and included in the model and adjusted for potential sources of heterogeneity. Pooled effica typical rFVIIa and APCC regimens were estimated. A total fo 17 studies, collectively reporting on more than 2,000 bleeds, were included. Medication type combined with dosage was the only significant explanatory parameter. Th predicts that a typical regimen of 90 microg kg(-1) rFVII repeated every 3 hours if needed results in cumulative join resolution of 66 %, 88 % and 95 % after 12, 24 and 36 hours, respectively. In comparison, a typical regimen of 75 APCC repeated every 12 hours if needed results in cumulative joint bleed resolution of 39 %, 62 % and 76 %, resp These differences were statistically significant and were also robust in sensitivity analyses. This analysis suggeste typical rFVIIa regimen will resolve joint bleeds more effectively than a typical APCC regimen after 12, 24 and 36 ho http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014 Clotting Factors Page 12 of31 Congenital factor XIII deficiency is rare and affects 1 out of every 3 to 5 million people in the United States. It is as with a tendency for severe bleeding, a risk for spontaneous abortion, and a high rate of spontaneous intra-cranial hemorrhage. The clinical severity of this deficiency requires regular prophylaxis from the time of diagnosis. Accor replacement material can be infused at intervals as long as every 20 to 30 days. Three types of factor XIII-contain are available: fresh-frozen plasma (FFP; preferably virus-inactivated), cryoprecipitate, and a pasteurized plasma co (Fibrogammin-P, Dade-Behring). The pasteurized concentrate and virus-inactivated FFP, when available, are pref cryoprecipitate (Lovejoy et al, 2006; Mannucci et al, 2010). On February 17, 2011, the FDA approved Corifact (factor XIII concentrate [Human]), derived from the pooled plasm healthy donors, via orphan-drug designation for the prevention of bleeding in people with congenital factor XIII defi FDA approved Corifact based on results of a clinical study of 14 people, including children, with congenital factor X deficiency. The most common side effects observed were hypersensitivity reactions (allergy, rash, pruritus, and er chills, fever, arthralgia, headache, elevated thrombin-antithrombin levels, and an increase in hepatic enzymes. It p can cause adverse events from abnormal clotting if doses higher than the labeled dose are given to patients. The U.S. Food and Drug Administration approved recombinant coagulation factor XIII A-subunit (Tretten, Novo No use in the routine prevention of bleeding in adults and children who have a congenital Factor XIII A-subunit deficie received orphan-drug designation for this use by the FDA because it is intended for treatment of a rare disease or condition. Congenital Factor XIII deficiency is a rare genetic disorder. Factor XIII is composed of two subunits, A an XIII deficiency is usually caused by a deficiency of the A-subunit. Tretten is a recombinant analogue of the human Factor XIII A-subunit that is produced in yeast cells and then furth It is a sterile freeze-dried-powder to be reconstituted with diluent and injected intravenously. Tretten can be admini physician or be self-administered. The effectiveness of coagulation factor XIII A-subunit was studied in 77 patients congenital Factor XIII Asubunit deficiency. The study found that coagulation factor XIII A-subunit was effective in p bleeding in 90 percent of the patients when given monthly. Among the side effects reported in this study were hea in the extremities and pain at injection site. No individual in the trial developed abnormal clots. The FDA-approved labeling for Tretten states that the dose for routine prophylaxis for bleeding in patients with con factor XIII (FXIII) A-subunit deficiency is 35 international units (IU) per kilogram body weight once monthly to achiev trough level of FXIII activity at or above 10% using a validated assay. The labeling states that dose adjustment sho considered if adequate coverage is not achieved with the recommended 35 IU/kg dose. Logan et al (2011) stated that rFVIIa is increasingly used for off-label indications. In a retrospective database analy investigators estimated patterns of off-label rFVIIa use in U.S. hospitals. Data were extracted from the Premier Pe database (Premier, Charlotte, NC), which contains discharge records from a sample of academic and non-academ hospitals. A total of 12,644 hospitalizations for patients who received rFVIIa during a hospital stay were analyzed. diagnoses and patient dispositions from 1 January 2000 to 31 December 2008 were reviewed. Statistical weights f hospital were used to provide national estimates of rFVIIa use. From 2000 to 2008, off-label use of rFVIIa in hospi increased more than 140-fold, such that in 2008, 97 % (95 % CI: 96 % to 98 %) of 18,311 in-hospital uses were off contrast, in-hospital use for hemophilia increased less than 4-fold and accounted for 2.7 % (CI: 1.9 % to 3.5 %) of u 2008. Adult and pediatric cardiovascular surgery (29 % CI: 21 % to 33 %), body and brain trauma (29 % CI: 19 % and intracranial hemorrhage (11 % CI: 7.7 % to 14 %) were the most common indications for rFVIIa use. Across a indications, inhospital mortality was 27 % (CI: 19 % to 34 %) and 43 % (CI: 26 % to 59 %) of patients were discha home. The authors concluded that off-label use of rFVIIa in the hospital setting far exceeds use for approved indic These patterns raise concern about the application of rFVIIa to conditions for which strong supporting evidence is l Yank et al (2011) evaluated the benefits and harms of rFVIIa use for 5 off-label, in-hospital indications: (i) cardiac s ICH, (iii) liver transplantation, (iv) prostatectomy, and (v) trauma. A total of 10 databases (including PubMed, EMBA the Cochrane Library) queried from inception through December 2010. Articles published in English were analyzed reviewers independently screened titles and abstracts to identify clinical use of rFVIIa for the selected indications a http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014 Clotting Factors Page 13 of31 all randomized, controlled trials (RCTs) and observational studies for full-text review. Two reviewers independently study characteristics and rated study quality and indication-wide strength of evidence. A total of 16 RCTs, 26 com observational studies, and 22 non-comparative observational studies met inclusion criteria. Identified comparators limited to placebo (RCTs) or usual care (observational studies). For ICH, mortality was not improved with rFVIIa u range of doses. Arterial thrombo-embolism was increased with medium-dose rFVIIa use (risk difference [RD], 0.03 0.01 to 0.06]) and high-dose rFVIIa use (RD, 0.06 [CI: 0.01 to 0.11]). For adult cardiac surgery, there was no mort difference, but there was an increased risk for thrombo-embolism (RD, 0.05 [CI: 0.01 to 0.10]) with rFVIIa. For bod there were no differences in mortality or thrombo-embolism, but there was a reduced risk for the acute respiratory syndrome (RD, -0.05 [CI: -0.02 to -0.08]). Mortality was higher in observational studies than in RCTs. The authors that limited available evidence for 5 off-label indications suggested no mortality reduction with rFVIIa use. For som indications, it increases thrombo-embolism. In an editorial that accompanied the afore-mentioned studies, Avorn and Kesselheim (2011) stated that overall, Ya workers found no evidence that rFVIIa reduced mortality for any off-label use; however, it did increase the risk for t embolism. Their findings are compatible with other recent studies. The editorialists noted that "[a]llowing physicia to choose medications is appealing, but not when it results in unhelpful, dangerous, and costly decisions. With suc compelling data in place about the runaway use, uselessness, and risk for this expensive treatment, what can be d reduce it? First, if evidence should emerge that the manufacturer played a role in building a market for the unautho increasingly implausible prescribing of its product, both civil and criminal responses will probably be brought to bea occurred for many other instances of corporate-sponsored drug misuse. Second, rFVIIa is used in hospitals, which providing organizational oversight to protect patients, as well as the institutions’ own pharmacy budgets. In hospit such use continues, existing quality assurance, patient safety, and riskmanagement groups will surely want to look these practices. Although off-label prescribing by physicians is not illegal, physicians who persist in such use in th clear evidence of inutility and harm could be subject to civil action by the affected patients or their heirs". Koncar et al (2011) examined the influence of rFVIIa on the treatment of intractable peri-operative bleeding in vasc when conventional hemostatic measures are inadequate. There were 2 groups of patients: the NovoSeven group 10 patients with ruptured abdominal aortic aneurysms (RAAAs) and 14 patients operated on due to thoraco-abdom aneurysms (TAAAs); the control group (group C), 14 patients with RAAAs and 17 patients with TAAAs. All patient intractable hemorrhage refractory to conventional hemostatic measures, while patients from group N were addition with rFVIIa. Postoperative blood loss was significantly lower in group N treated with rFVII (p < 0.0001). Post-oper administration of packed red blood cells, fresh frozen plasma, and platelets was lower in patients from group N, (p Successful hemorrhage arrest was reported in 21 patients (87.5 %) treated with rFVIIa, and in 9 patients (29.03 %) (p < 0.001). Thirty-day mortality in these 2 groups significantly differed. The mortality rate was 12.5 % (3 patients) and 80.65 % (25 patients) in group C (p < 0.0001). The authors concluded that these findings suggested that rFVI a role in controlling the intractable peri-operative and postoperative bleeding in surgical patients undergoing a rep RAAAs and TAAAs. They stated that prospective randomized trials are necessary to further confirm the efficacy a effectiveness of rFVIIa in these patients. Witmer et al (2011) described the off-label use of rFVIIa in tertiary care pediatric hospitals across the United States assess thrombotic events. A retrospective multi-center cohort study using the Pediatric Health Information System administrative database was performed. Children 18 years of age or younger who received rFVIIa between 2000 a were included. A label admission was defined as an admission with an International Classification of Diseases dia code for hemophilia or factor VII deficiency; admissions without these codes were classified as off-label. There we rFVIIa admissions, representing 3,764 individual subjects; 74 % (3,655) of the admissions were off-label. There wa increase in the annual rate of off-label admissions from 2000 to 2007 (from 2 to 20.8 per 10,000 hospital admissio 0.001). The mortality rate in the off-label group was 34 % (1,258/3,655). Thrombotic events occurred in 10.9 % (3 the offlabel admissions. The authors concluded that off-label use of rFVIIa in hospitalized children is increasing ra despite the absence of adequate clinical trials demonstrating safety and efficacy. Thrombotic events are common mortality is high among patients receiving off-label rFVIIa. They stated that further studies are warranted to determ these adverse events are attributable to rFVIIa. http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014 Page 14 of31 Clotting Factors In a Cochrane review, Lin et al (2011) evaluated the effectiveness of rFVIIa when used therapeutically to control ac bleeding, or prophylactically to prevent (excessive) bleeding in patients without hemophilia. These investigators se Cochrane Injuries Group Specialized Register, CENTRAL, MEDLINE, EMBASE and other specialized databases u February 2009. Randomized controlled trials comparing rFVIIa with placebo, or one dose of rFVIIa with another, i population (except hemophilia) were included in this study. Outcomes were mortality, blood loss or control of bleed transfusion requirements, number of patients transfused and thrombo-embolic adverse events. Two authors indep assessed potentially relevant studies for inclusion, extracted data and examined risk of bias. They considered prop and therapeutic rFVIIa studies separately. A total of 25 RCTs were included: 24 were placebo-controlled double-b and 1 compared different doses of rFVIIa. Fourteen trials involving 1,137 participants examined the prophylactic u 713 received rFVIIa. There was no evidence of mortality benefit (RR 1.06; 95 % CI: 0.50 to 2.24). There was decr blood loss (WMD -272 ml; 95 % CI: -399 to -146) and decreased red cell transfusion requirements (WMD -243 ml; 393 to -92) with rFVIIa treatment; however these values were likely over-estimated due to the inability to incorporat trials showing no difference of rFVIIa treatment compared to placebo. There was a trend in favor of rFVIIa in the n participants transfused (RR 0.91; 95 % CI: 0.82 to 1.02). But there was a trend against rFVIIa with respect to thro adverse events (RR 1.32; 95 % CI: 0.84 to 2.06). Eleven trials involving 2,366 participants examined the therapeu rFVIIa; 1,507 received rFVIIa. There were no outcomes where any observed advantage, or disadvantage, of rFVII placebo could not have been observed by chance alone. There was a trend in favor of rFVIIa for reducing mortality 95 % CI: 0.77 to 1.03). However, there was a trend against rFVIIa for increased thrombo-embolic adverse events 5% CI: 0.93 to 1.58). The authors concluded that the effectiveness of rFVIIa as a more general hemostatic drug, e prophylactically or therapeutically, remains unproven. They stated that the use of rFVIIa outside its current license indications should be restricted to clinical trials. Wilate is a human plasma-derived, sterile, purified, double virus inactivated von Willebrand factor/coagulation facto Complex (human). It is indicated for the treatment of spontaneous and/or trauma-induced bleeding episodes in ind with severe von Willebrand disease (VWD) or individuals with mild or moderate VWD when there is failure, contrai intolerance to desmopressin. It is not indicated for individuals with hemophilia A; and for the prophylaxis of sponta bleeding episodes, or the prevention of excessive bleeding during and after surgery in individuals with VWD. The m common adverse reactions to treatment with Wilate in patients with VWD have been urticaria and dizziness. The adverse reactions to treatment with Wilate in patients with VWD have been hypersensitivity reactions. An UpToDate review on “Factor VIII and factor IX inhibitors in patients with hemophilia” (Hoots and Shapiro, 2012a mention the use of von Willebrand factor/coagulation factor VIII complex (Wilate). Moreover, an UpToDate review “Treatment of hemophilia” (Hoots and Shapiro, 2012b) states that “Humate P, one of these products, contains both and von Willebrand factor, and is mainly used for the treatment of von Willebrand disease. It is not a preferred age treatment of patients with hemophilia A who do not have inhibitors”. Franchini et al (2013) summarized the current knowledge on treatment strategies for hemophilia B, focusing on rec FIX (rFIX) products either clinically used or in development. There is only 1 rFIX product that is licensed to treat he patients. From the analysis of the literature data presented in this review, the authors concluded that this rFIX pro demonstrated an excellent safety profile and excellent clinical effectiveness for halting and preventing bleeds in he patients. While prophylaxis has emerged as the best therapeutic strategy for such patients because of its ability to hemophilic arthropathy, and to improve patients' quality of life, the pharmacokinetically tailored dosing of rFIX is an point when planning hemophilia B treatment, as it allows optimization of the factor concentrate usage. Windyga et al (2014) stated that BAX326 is a rFIX manufactured without the addition of any materials of human or origin, and with 2 viral inactivation steps (solvent/detergent treatment and 15 nm nano-filtration). The aim of this pr trial was to investigate the pharmacokinetics, hemostatic efficacy and safety of BAX326 in previously treated patie to 65 years with severe or moderately severe hemophilia B. BAX326 was safe and well-tolerated in all 73 treated s adverse events considered related to treatment (2.7 % incidence, all non-serious) were transient and mild, and no hypersensitivity reactions, inhibitor formation or thrombotic events were observed. Pharmacokinetic (PK) equivale between BAX326 and a licensed rFIX was confirmed in terms of the ratio of geometric mean AUC0-72 h per dose. http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014 Clotting Factors Page 15 of31 weekly prophylaxis [mean duration 6.2 (+/- 0.7) months; 1.8 (+/- 0.1) infusions per week, 49.5 (+/- 4.8) IU kg-1 per was effective in preventing bleeding episodes, with a significantly lower (79 %, p < 0.001) annualized bleed rate (4 compared to an on-demand treatment in a historical control group (20.0); 24 of 56 subjects on prophylaxis (43 %) d throughout the study observation period. Of 249 total acute bleeds, 211 (84.7 %) were controlled with 1 to 2 infusi BAX326. Hemostatic efficacy at resolution of bleed was rated excellent or good in 96.0 % of all treated bleeding ep The authors concluded that these findings indicated that BAX326 is safe and effectives in treating bleeds and routi prophylaxis in patients aged 12 years and older with hemophilia B. On June 27, 2013, the FDA approved Rixubis [Coagulation Factor IX (Recombinant)] for use in people with hemop are 16 years of age and older. Rixubis is indicated for the control and prevention of bleeding episodes, peri-operat extending from the time of hospitalization for surgery to the time of discharge) management, and routine use to pre reduce the frequency of bleeding episodes (prophylaxis). Rixubis is a purified protein produced by recombinant DN technology. It does not contain human or animal proteins. It is supplied in single-use vials of freeze-dried powder administered by intravenous injection after reconstitution with sterile water for injection. When used for the routine of bleeding episodes, it is administered twiceweekly. The effectiveness of Rixubis was evaluated in a multi-center which a total of 73 male patients between 12 and 65 years of age received Rixubis for routine prophylaxis or as ne response to symptoms of bleeding (on-demand). Overall, patients in the prophylaxis study had a 75 % lower annu rate when compared to patients who have historically received ondemand treatment. An additional study in a ped population is currently ongoing. Although serious side effects including anaphylaxis can occur, the most common observed in patients in clinical studies were dysgeusia, pain in an extremity, and atypical blood test results. An UpToDate review on “Treatment of hemophilia” (Hoots, 2013) states that “Recombinant human factor IX has be genetically engineered by insertion of the human factor IX gene into a Chinese hamster ovary cell line. It has been be safe and effective in the treatment of patients with previously treated and previously untreated hemophilia B, wit of 16 to 17 hours. The product has no added albumin, giving it a theoretical advantage over plasma-derived conce The volume of distribution of recombinant factor IX is larger than that for plasma-derived factor IX, with a more pron increase in infants and children. Available products include BeneFIX and Rixubis, which was licensed by the US F Drug Administration in 2013”. The U.S. Food and Drug Administration approved Alprolix (Biogen Idec, Cambridge, MA), Coagulation Factor IX (Recombinant), Fc Fusion Protein, for use in adults and children who have hemophilia B to help control and preven episodes, manage bleeding during surgical procedures, and prevent or reduce the frequency of bleeding episodes (prophylaxis). Alprolix is designed to require less frequent injections when used to prevent or reduce the frequency Alprolix consists of the Factor IX molecule linked to a protein fragment, Fc, to make the product last longer in circul Alprolix received orphan-drug designation for this use by the FDA because it is intended for treatment of a rare dis condition. The safety and efficacy of Alprolix were evaluated in a multi-center clinical trial that compared each of two prophyla treatment regimens to on-demand treatment. Powell, et al. (2013) conducted a phase 3, nonrandomized, open-lab the safety, efficacy, and pharmacokinetics of Alprolix for prophylaxis, treatment of bleeding, and perioperative hem 123 previously treated male patients. All participants were 12 years of age or older and had severe hemophilia B (e factor IX level of ≤2 IU per deciliter, or ≤2% of normal levels). The study included four treatment groups: group 1 re weekly doseadjusted prophylaxis (50 IU of Alprolix per kilogram of body weight to start), group 2 received interval- prophylaxis (100 IU per kilogram every 10 days to start), group 3 received treatment as needed for bleeding episod 100 IU per kilogram), and group 4 received treatment in the perioperative period. A subgroup of group 1 underwen comparative sequential pharmacokinetic assessments of recombinant factor IX and Alprolix. The primary efficacy was the annualized bleeding rate, and safety end points included the development of inhibitors and adverse events compared with recombinant factor IX, Alprolix exhibited a prolonged terminal half-life (82.1 hours) (P<0.001). The annualized bleeding rates in groups 1, 2, and 3 were 3.0, 1.4, and 17.7, respectively. In group 2, 53.8% of participa dosing intervals of 14 days or more during the last 3 months of the study. In groups 1, 2 and 3, 90.4% of bleeding resolved after one injection. Hemostasis was rated as excellent or good during all major surgeries. No inhibitors w http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014 Clotting Factors Page 16 of31 detected in any participants receiving Alprolix; in groups 1, 2, and 3, 73.9% of participants had at least one adverse serious adverse events occurred in 10.9% of participants. These events were mostly consistent with those expecte general population of patients with hemophilia. The authors concluded that prophylactic Alprolix, administered ever weeks, resulted in low annualized bleeding rates in patients with hemophilia B. One IU of Alprilox per kg body weight increases the circulating level of Factor IX by 1%. The recommended dose f prophylaxis is 50 IU/kg once weekly or 100 IU/kg once every 10 days, with dosing regimen adjusted based upon in response. For control of minor and moderate bleeding episodes (for example, uncomplicated hemarthoses, superf bleeding (except iliopsoas) without neurovascular compromise, superficial soft tissue bleeding, bleeding of mucous membranes) requires a circulating factor IX level of 30 to 60 IU/dL or 30 to 60 percent of normal). For control of ma (for example, iliopsoas and deep muscle bleeding with neurovascular injury, or substantial blood loss; pharyngea, retropharyngeal, retroperitoneal or CNS bleeding) requires a circulating factor IX level of 80 to 100 IU/dL or 80 to 1 of normal. For minor surgery (including uncomplicated dental extraction), the required circulating Factor IX level is IU/dL or 50 to 80% normal. For major surgery, the required circulating Factor IX level is 60 to 100 IU/dL or 60 to 10 normal. Patients receiving chronic anti-coagulation therapy with warfarin and other vitamin K antagonist (VKA) anti-coagula prevent blood clotting in conditions such as atrial fibrillation or the presence of an artificial heart valve sometimes d acute bleeding. Hickey et al (2013) determined adverse event frequency after urgent reversal with frozen plasma v prothrombin complex concentrate (PCC) -- Octaplex. This natural before-after retrospective cohort study in 2 tertia emergency departments compared anti-coagulation reversal with frozen plasma (September 2006 to August 2008) Octaplex (September 2008 to August 2010), without other system changes. These researchers included adult pat warfarin with an international normalized ratio (INR) of greater than or equal to 1.5 who received frozen plasma or O The primary outcome was serious adverse events (death, ischemic stroke, myocardial infarction, heart failure, ven thromboembolism, or peripheral arterial thromboembolism) within 7 days. Secondary outcomes included time to IN hospital length of stay, and red blood cells transfused within 48 hours. They included 149 patients receiving frozen and 165 receiving Octaplex. The incidence of serious adverse events for the frozen plasma group was 19.5 % com 9.7 % for the Octaplex group (p = 0.014; relative risk, 2.0; 95 % CI: 1.1 to 3.5). This remained significant after adju baseline history and reason for treatment (p = 0.038; adjusted relative risk, 1.85; 95 % CI: 1.03 to 3.3) in multi-varia regression analysis. Median INR reversal was 11.8 hours with frozen plasma and 5.7 hours with Octaplex (p < 0.0 red cell transfusion was 3.2 with frozen plasma and 1.4 with Octaplex (p < 0.0001). The authors concluded that Oc urgent reversal of warfarin resulted in faster reversal and lower red cell transfusion requirement with fewer adverse than frozen plasma. Hanke et al (2013) noted that the rapid reversal of the effects of VKA is often required in cases of emergency surge threatening bleeding, or during bleeding associated with high morbidity and mortality such as intra-cranial hemorrh Increasingly, 4-factor PCCs (4F-PCC) containing high and well-balanced concentrations of vitamin K-dependent c factors are recommended for emergency oral anti-coagulation reversal. Both the safety and effectiveness of such are currently in focus, and their administration is now expanding into the critical care setting for the treatment of life bleeding and coagulopathy resulting either peri-operatively or in cases of acute trauma. After 15 years of clinical u of a pharmaco-vigilance report (February 1996 to March 2012) relating to the 4F-PCC Beriplex P/N (CSL Behring, Germany) were analyzed and were presented here. Furthermore, a review of the literature with regard to the safet effectiveness of 4F-PCCs was performed. Since receiving marketing authorization (February 21, 1996), approxima 647,250 standard applications of Beriplex P/N have taken place. During this time, 21 thromboembolic events judg possibly related to Beriplex P/N administration have been reported, while no incidences of viral transmission or hep induced thrombocytopenia were documented. The low risk of thromboembolic events reported during the observa (1 in approximately 31,000) is in line with the incidence observed with other 4F-PCCs. The authors concluded that 4F-PCCs have proven to be well-tolerated and highly effective in the rapid reversal of VKA. Sarode et al (2013) performed a prospective clinical trial to compare non-activated 4F-PCC with plasma for urgent reversal. In this phase IIIb, multi-center, open-label, non-inferiority trial, non-surgical patients were randomized to http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014 Clotting Factors Page 17 of31 (containing coagulation factors II, VII, IX, and X and proteins C and S) or plasma. Primary analyses examined whe PCC was non-inferior to plasma for the co-primary end-points of 24-hour hemostatic efficacy from start of infusion correction (less than or equal to 1.3) at 0.5 hour after end of infusion. The intention-to-treat efficacy population com patients (4F-PCC, n = 98; plasma, n = 104). Median (range) baseline INR was 3.90 (1.8 to 20.0) for the 4F-PCC g 3.60 (1.9 to 38.9) for the plasma group. Effective hemostasis was achieved in 72.4 % of patients receiving 4F-PC 65.4 % receiving plasma, demonstrating non-inferiority (difference, 7.1 % [95 % CI: -5.8 to 19.9]). Rapid INR reduc achieved in 62.2 % of patients receiving 4F-PCC versus 9.6 % receiving plasma, demonstrating 4F-PCC superiorit (difference, 52.6 % [95 % CI: 39.4 to 65.9]). Assessed coagulation factors were higher in the 4F-PCC group than i plasma group from 0.5 to 3 hours after infusion start (p < 0.02). The safety profile (adverse events, serious advers thromboembolic events, and deaths) was similar between groups; 66 of 103 (4F-PCC group) and 71 of 109 (plasm patients experienced greater than or equal to 1 adverse event. The authors concluded that 4F-PCC is an effective to plasma for urgent reversal of VKA therapy in major bleeding events, as demonstrated by clinical assessments of and laboratory measurements of INR and factor levels. On April 29, 2013, the FDA approved Kcentra (prothrombin complex concentrate, human) for the urgent reversal o coagulation factor deficiency induced by VKA (e.g., warfarin) therapy in adult patients with acute major bleeding. K not indicated for urgent reversal of VKA anti-coagulation in patients without acute major bleeding. The FDA approv Kcentra was based on a study of 216 patients who had been receiving VKA anti-coagulation and who had acute m bleeding along with a clotting test value indicative of anti-coagulant use. Kcentra was demonstrated to be similar t terms of the ability to stop acute major bleeding. Kcentra is made from the pooled plasma of healthy donors. It is p in a way to minimize the risk of transmitting viral and other diseases. Ferreira and DeLosSantos (2013) stated that PCC is an inactivated concentrate of factors II, IX, and X, with variab of factor VII. Guidelines recommend the use of PCC in the setting of life-threatening bleeds, but little is known on t effective dosing strategies and how the presenting international normalized ratio affects response to therapy. Thes investigators high-lighted available data on monitoring techniques, address shortcomings of currently available dat reversal of life-threatening and critical bleeds with PCC, and how this product compares to other therapeutic option critically ill patients. Prothrombin complex concentrate has been identified as a potential therapy for critically bleed but patient-specific factors, product availability, and current data should weigh the decision to use it. Most data exi patients experiencing VKA-induced bleeding, more specifically, those with intra-cranial hemorrhage. Prothrombin c concentrate has also been studied in trauma-induced hemorrhage; however, it remains controversial, as its potenti have the abilities to become flaws in this setting. The authors concluded that health care professionals must rema the differences in products and interpret how 3- versus 4-factor products may affect patients, and interpret literatur accordingly. The clinician must be cognizant of how to progress when treating a bleeding patient, propose a suppo scheme, and address the need for appropriate factor VII supplementation. At this point, PCC cannot be recomme -line therapy in patients with traumatic hemorrhage, and should be reserved for refractory bleeding until more data available. The National Institute for Health and Clinical Excellence’s clinical guideline on “Acute upper gastrointestinal bleedin Management” (NICE, 2012) recommended PCC to patients who are taking warfarin and actively bleeding. Dewhirst (2013) performed a short-cut review to establish whether rFVIIa improves survival and functional outcom spontaneous intracranial hemorrhage. A total of 92 papers were found using the reported searches, of which 2 pre best evidence to answer the clinical question. The author, date and country of publication, patient group studied, s relevant outcomes, results and study weaknesses of these best papers were tabulated. The authors concluded tha evidence does not support the use of rFVIIa in acute spontaneous intracranial hemorrhage. Awad and Cocchio (2013) stated that PCC products are emerging as alternative strategies for reversing anticoagu pharmacotherapy. Factor eight inhibitor bypassing activity (FEIBA, or anti-inhibitor coagulant complex) is an activa (aPCC). Although FEIBA is approved by the FDA to control spontaneous bleeding episodes and to prevent bleedin surgical interventions in hemophilia A and hemophilia B patients with inhibitors to factor VIII, recent data have sug the product may be used off-label as an anticoagulant-reversal agent. These researchers evaluated the safety and http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014 Clotting Factors Page 18 of31 effectiveness of aPCC products in reversing anticoagulant pharmacotherapy. They searched online databases for language publications that discussed this topic. The EMBASE, MEDLINE, and International Pharmaceutical Abstr databases were used. These researchers evaluated all articles published in the English language identified from th sources. They included studies conducted in human subjects and in in-vitro and in-vivo models in this review. Cur published evidence suggested that the use of an aPCC, compared with fresh-frozen plasma, is associated with a s faster correction of supra-therapeutic INRs secondary to warfarin therapy. Conflicting evidence exists regarding th aPCCs to reverse the prolonged bleeding times caused by the anticoagulant agents including dabigatran etexilate rivaroxaban (Xarelto), apixaban (Eliquis), and fondaparinux (Arixtra). The authors concluded that the theoretical ri thrombosis associated with PCC products must be carefully considered before they are administered to patients w coagulation therapy. The use of aPCCs to reverse the anticoagulant effects of warfarin, dabigatran, or rivaroxaban limited because of the lack of safety and effectiveness data in humans. Moreover, the safety of aPCCs in off-label has not been adequately assessed. Stewart and Pettit (2013) summarized their experiences with FEIBA for the reversal of warfarin-related bleeding in community hospital. A protocol was put in place in March of 2011, which outlined the use of FEIBA for the emerge of warfarin-related coagulopathy. A fixed low-dose was given based on INR. For an INR less than 5.0, 500 U of F administered’ for an INR greater than or equal to 5.0, 1,000 U of FEIBA was given. Intravenous vitamin K was give concurrently regardless of INR. A total of 16 patients were treated with FEIBA per the protocol. Average patient a years. Intracranial hemorrhage was the most common indication for reversal. Mean pre-treatment INR was 3.56 ( mean post-treatment INR was 1.16 (1.01 to 1.32). Two of the patients required a second 500-U dose, per the proto INR that had not yet normalized. Bleeding appeared clinically controlled in 93 % of cases; 87 % of patients survive discharge. There were no signs or symptoms of thrombosis in any of the cases. The authors concluded that emer reversal of warfarin utilizing a fixed low-dose of FEIBA appears to be effective, consistent, and safe. Moreover, th that further comparator studies with other reversal agents are needed. Cabral and colleagues (2013) evaluated the safety and effectiveness of a PCC-based protocol in patients with warf associated ICH, sub-dural hematoma (SDH), or sub-arachnoid hemorrhage (SAH). This was a retrospective case- review of patients treated with an institution-approved warfarin reversal protocol. Patients with ICH and known war with an INR greater than 1.4 received FFP, vitamin K (phytonadione), and weight-based, 3-factor PCC (Profilnine( based on the initial INR. Demographic and clinical information, the degree of and time to INR normalization, and a events were recorded. The 30 study patients included 19 with primary ICH, 7 with SDH, and 4 with SAH. The me 72.8 (± 11) years, including 11 (37 %) patients greater than or equal to 80 years old. The median presenting INR w (IQR 2 to 3.3) and post-treatment INR was 1.4 (IQR 1.3 to 1.5, Z score 6.4, p < 0.001). Median time from PCC ad to the first follow-up INR was 95 (IQR 50 to 140) mins. No patient's INR increased by more than 0.3 over 72 hrs. N patients (30 %) underwent neurosurgical procedures after PCC administration and no procedure-related bleeding was noted. Adverse events included 3 instances of early hematoma expansion, 1 ischemic stroke in a patient with endocarditis on post-PCC day 1, 1 pulmonary embolism 5 weeks after PCC treatment, and 1 coronary in-stent thro days after PCC treatment. Six patients died prior to hospital discharge of anticipated complications of their initial e none from identifiable thrombotic complications of PCC. The authors concluded that a 3-factor PCC preparation ( SD), administered with FFP and vitamin K to patients with acute warfarin-associated intracranial bleeding is a reas approach to urgent warfarin reversal. Moreover, they stated that randomized, prospective trials are needed to verif and clinical effectiveness of PCC administration in this population. Song et al (2013) reported their initial experience with the PCC FEIBA for the rescue treatment of coagulopathy an threatening bleeding after cardiac surgery. A total of 25 patients who underwent cardiac surgery with coagulopath threatening bleeding refractory to conventional treatment received FEIBA as rescue therapy at the authors’ instituti cohort represented approximately 2 % of patients undergoing cardiac surgery in the authors’ university-based prac the study. The patients were at high risk for post-operative coagulopathy with nearly all patients having at least 2 r for this. Aortic root replacement (Bentall or valve-sparing procedure) and heart transplant with or without left ventri device explant were the most common procedures. The mean FEIBA dose was 2,154 units. The need for fresh fro and platelet transfusion decreased significantly after FEIBA administration (p = 0.0001 and p < 0.0001). The mean http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014 Clotting Factors Page 19 of31 decreased from 1.58 to 1.13 (p < 0.0001). Clinical outcomes were excellent. No patient returned to the operating exploration. There was no hospital mortality and all patients were discharged home. One patient who had a centra transvenous pacemaker developed an upper extremity DVT. The authors concluded that their initial experience w administration for the rescue treatment of post-operative coagulopathy and life-threatening bleeding has been favo Moreover, they stated that further studies are indicated to confirm its safety and effectiveness; and determine spe indications for its use in patients undergoing cardiac surgery. The U.S. Food and Drug Administration (FDA) has approved Eloctate [Antihemophilic Factor (Recombinant), Fc Fu Protein] for the control and prevention of bleeding episodes, perioperative (surgical) management and routine prop adults and children with hemophilia A (Biogen Idec, 2014). Eloctate reduces the frequency of bleeding episodes wi prophylactic infusions every three to five days, offering people with hemophilia A the potential to extend the interva prophylactic infusions. Eloctate was developed by fusing B-domain deleted factor VIII to the Fc portion of immunog subclass 1 (IgG1) which enables Eloctate to prolong the time the therapy remains in the body. The recommended starting prophylactic regimen for Eloctate is 50 IU/kg every four days. Based on clinical respon regimen may be adjusted in the range of 25 to 65 IU/kg and every three to five days (Biogen Idec., 2014). In clinical trials, Eloctate was effective for both routine prophylaxis and to treat acute bleeding episodes with a favo and tolerability profile (Biogen Idec, 2014). The approval of Eloctate is based on results from the global, Phase 3 A clinical study, as well as interim pharmacokinetic and safety data from the Phase 3 Kids A-LONG study. The A-LONG study was an open-label, multi-center study that examined the efficacy, safety and pharmacokinetics in 165 previously treated males 12 years of age and older with severe hemophilia A (Biogen Idec, 2014). Results s adults and adolescents with severe hemophilia A achieved a statistically significant reduction of bleeding episodes the study’s prophylaxis arms, relative to the on-demand treatment arm. In addition, 98 percent of bleeding episode controlled with one or two Elocttate infusions. The study evaluated individualized and weekly prophylaxis to reduce or prevent bleeding episodes, and on-deman treat bleeding episodes (Biogen Idec, 2014). In the individualized arm, each study participant started on a twice-we regimen. Participants’ pharmacokinetic parameters were used to guide adjustments to dosing interval (every three days), and dose (25 to 65 IU/kg) to target a minimum factor VIII level of 1 to 3 IU/dL or higher as needed to mainta breakthrough bleeding episodes. In the study, the dose in the weekly prophylaxis arm was 65 IU/ kg/week. The ov annualized bleeding rates (ABR), or projected number of bleeding episodes per year, reported in the study were 1. individualized prophylaxis arm, 3.6 for the weekly prophylaxis arm and 33.6 for the on-demand arm. No participants in the A-LONG study developed inhibitors to Eloctate (Biogen Idec, 2014). One participant had a tra positive neutralizing antibody test result, which was not confirmed upon repeat testing. There were no reports of se vascular clots or serious allergic reactions. Across the routine prophylaxis and on-demand therapy arms, adverse r were reported in 5.5 percent of participants. Adverse reactions included arthralgia, malaise, upper abdominal pain, abdominal pain, angiopathy, bradycardia, chest pain, cough, dizziness, dysgeusia, cold and heat intolerance, head hypertension, joint swelling, myalgia, procedural hypotension and rash. Each event occurred in two or fewer study Two participants were withdrawn from the study due to adverse reactions: one participant due to rash and one due arthralgia. The pediatric indication for Eloctate is supported by interim safety and pharmacokinetic results in 38 boys ages tw old from the Phase 3 Kids A-LONG study (Biogen Idec, 2014). These data showed that Eloctate was generally wel and no inhibitors were detected. The relative increase in half-life seen with Eloctate was consistent with findings in adolescents. In comparison with adolescents and adults, children two to five years old have a shorter half-life and h clearance of hemophilic factors (adjusted for body weight); therefore, a higher dose or more frequent dosing may b this age group. In April 2014, Biogen Idec and Swedish Orphan Biovitrum (Sobi) reported positive top-line results fr completed Kids A-LONG study, which confirmed and expanded upon the interim data. Common adverse reactions of greater than or equal to 1 percent) reported in the A-LONG study were arthralgia and malaise. http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014 Page 20 of31 Clotting Factors CPT Codes / HCPCS Codes / ICD-9 Codes Other CPT codes related to the CPB: 85002 85335 85610 - 85611 85730 - 85732 96365 - 96379 99601 + 99602 Other HCPCS codes related to the CPB: S9345 Home infusion therapy, antihemophilic agent infusion therapy (e.g., factor VIII); administ services, professional pharmacy services, care coordination, and all necessary supplies equipment (drugs and nursing visits coded separately), per diem Anti-hemophilic factor (factor VIII), factor IX and Humate-P or Alphanate: Other CPT codes related to the CPB: 85240 85244 85245 85246 85247 85250 HCPCS codes covered if selection criteria are met: C9133 Factor ix (antihemophilic factor, recombinant), rixibus, per i.u. J7180 Injection, factor XIII (antihemophilic factor, human), 1 I.U. J7183 Injection, Von Willebrand factor complex (human), wilate, 1 I.U. VWF:RCO J7185 Injection, Factor VIII (Antihemophilic Factor, recombinant) (Xyntha), per I.U. J7186 Injection, antihemophilic factor VIII / von Willebrand factor complex (human), per factor V J7187 Injection, von Willebrand factor complex (Humate-P), per IU vWF-RCO J7190 - J7192 Hemophilia clotting factor VIII http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014 Page 21 of31 Clotting Factors J7193 - J7195 Hemophilia clotting factor IX ICD-9 codes covered if selection criteria are met: 286.0 Congenital factor VIII disorder [Hemophilia A] 286.1 Congenital factor IX disorder [Hemophilia B] 286.4 von Willebrand's disease Recombinant factor VIII Fc fusion protein (Eloctate): No specific code ICD-9 codes covered if selection criteria are met: 286.0 Congenital factor VIII disorder [Hemophilia A] Recombinant factor VIIa (rFVIIa, NovoSeven): Other CPT codes related to the CPB: 85230 HCPCS codes covered if selection criteria are met: J7189 Factor VIIa (antihemophilic factor, recombinant) per 1 mcg Q2023 Injection, factor viii (antihemophilic factor, recombinant) (Xyntha), per i.u. ICD-9 codes covered if selection criteria are met: 286.0 Congenital factor VIII disorder [Hemophilia A] 286.1 Congenital factor IX disorder [Hemophilia B] 286.3 Congenital deficiency of other clotting factors 286.52 Acquired hemophilia 287.1 Qualitative platelet defects [Glanzmann's thrombasthenia with antibodies to glycoprotein and/or human leukocyte antigen (HLA), refractory to platelet infusions] ICD-9 codes not covered for indications listed in the CPB: 411.1 Intermediate coronary syndrome 411.81 Acute coronary occlusion without myocardial infarction 430 - 438.9 Cerebrovascular disease [including treatment of acute spontaneous intracerebral hemo unrelated to hemophilia] 456.0 - 456.21 Esophageal varices 555.0 - 555.9 Regional enteritis [Crohn's disease] 800.00 - 804.99 Fracture of skull [with intracranial bleeding] http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014 Page 22 of31 Clotting Factors 850.00 - 854.19 Intracranial injury, excluding those with skull fracture [with intracranial bleeding] 998.11 Hemorrhage complicating a procedure [not covered for peri-operative bleeding in vascu as well as trauma] Human anti-thrombin III (Thrombate III): Other CPT codes related to the CPB: 85300 85301 HCPCS codes covered if selection criteria are met: J7197 Anti-thrombin III (human) [Thrombate III] ICD-9 codes covered if selection criteria are met: 289.81 Primary hypercoagulable state [anti-thrombin III deficiency] [covered in connection with embolism, obstetrical procedures, or surgical procedures only] Recombinant factor IX fc fusion protein (rFIXFc) (Alprolix): No specific code ICD-9 codes covered if selection criteria are met: 286.1 Congenital factor IX disorder [Hemophilia B] Factor VIII inhibitor bypassing activity (FEIBA) anti-inhibitor coagulant complex: Other HCPCS codes related to the CPB: J7198 Antiinhibitor, per IU ICD-9 codes covered if selection criteria are met: 286.0 Congenital factor VIII disorder [Hemophilia A] 286.1 Congenital factor IX disorder [Hemophilia B] 286.2 Congenital factor XI deficiency 286.3 Congenital deficiency of other clotting factors [acquired inhibitors to Factors VIII, XII] 964.2 Poisoning by anticoagulants [not for use for reversal of anticoagulant-associated coagu rescue treatment of coagulopathy after cardiac surgery] 997.1 Cardiac complications [rescue treatment of coagulopathy after cardiac surgery] ICD-9 codes not covered for indications listed in the CPB: 286.52 Acquired hemophilia [non-hemophiliac persons with acquired inhibitors to factors VIII, X Corifact (factor XIII concentrate [Human]): http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014 Page 23 of31 Clotting Factors Other CPT codes related to the CPB: 85290 85291 Other HCPCS codes related to the CPB: J7198 Antiinhibitor, per IU J7199 Hemophilia clotting factor, not otherwise classified ICD-9 codes covered if selection criteria are met: 286.3 Congenital deficiency of other clotting factors [factor XIII] Recombinant coagulation factor XIII A-subunit (Tretten): No specific code HCPCS codes covered if selection criteria are met: C9134 Factor XIII (antihemophilic factor, recombinant), Tretten, per 10 I.U. ICD-9 codes covered if selection criteria are met: 286.3 Congenital deficiency of other clotting factors [congenital factor XIII A-subunit deficiency von Willebrand factor/coagulation factor VIII complex (Wilate®): HCPCS codes covered if selection criteria are met: J7183 Injection, Von Willebrand factor complex (human), wilate, 1 I.U. VWF:RCO Other HCPCS codes related to the CPB: J2597 Injection, desmopressin, acetate, per 1 mcg ICD-9 codes covered if selection criteria are met: 286.0 Congenital factor VIII disorder [not covered for Hemophilia A] 286.4 von Willebrand's disease [not covered for prophylaxis of spontaneous bleeding episode ICD-9 codes not covered for indications listed in the CPB: 674.30, 674.32, 674.34 Other complication of obstetrical surgical wounds [prevention of excessive bleeding dur after surgery in individuals with VWD] 998.11 Hemorrhage complicating a procedure [prevention of excessive bleeding during and aft in individuals with VWD] Prothrombin complex concentrate (human) (Kcentra): HCPCS codes covered if selection criteria are met: C9132 Prothrombin complex concentrate (human), Kcentra, per i.u. of Factor IX activity http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014 Page 24 of31 Clotting Factors ICD-9 codes covered if selection criteria are met: 286.7 Acquired coagulation factor deficiency E934.2 Anticoagulants causing adverse effect in therapeutic use [e.g. Warfarin therapy] The above policy is based on the following references: 1. United States Pharmacopeial Convention, Inc. (USPC). USP Dispensing Information. Volume I -- Drug Infor the Health Care Professional. 18th ed. Rockville, MD: USPC; 1998. 2. Mosby-Year Book, Inc. Mosby's GenRx: The Complete Reference for Generic and Brand Drugs. 8th ed. St. Mosby; 1998. 3. American Society of Health-System Pharmacists, Inc. American Hospital Formulary Service Drug Informatio Bethesda, MD: American Society of Health-System Pharmacists; 1998. 4. Medical Economics, Inc. Physicians' Desk Reference. 52nd ed. Montvale, NJ: Medical Economics Data Cor 1998. 5. Lusher JM. Considerations for current and future management of haemophilia and its complications. Haemo 1991;1:1-20. 6. DiMichele D. Hemophilia 1996: New approaches to an old disease. Pediatr Clin North Am. 1996;43:709-73 7. Association of Hemophilia Clinic Directors of Canada. Hemophilia and von Willebrand's disease: 1. Diagnos comprehensive care and assessment. CMAJ. 1995;153:19-25. 8. Bray GL, Comperts ED, Courter S, et al. A multicenter study of recombinant Factor VIII (Recombinate): Saf and 9. inhibitor risk in previously untreated patients with hemophilia A. Blood. 1994;83:2429-2435. Nilsson IM, Berntorp E, Löfqvist T, et al. Twenty-five years' experience of prophylactic treatment in severe h A and B. J Intern Med. 1992;232:25-32. 10. Carlsson M, Berntorp E, Bjorkman S, et al. Pharmacokinetic dosing in prophylactic treatment of hemophilia Haematol. 1993;31:247-252. 11. Brackmann HH, Eickhoff HJ, Oldenburg J, et al. Long-term therapy and on-demand treatment of children an 12. adolescents with severe haemophilia A: 12 years of experience. Haemostasis. 1992;22:251-258. Berntorp E, Nilsson IM. Use of a high-purity Factor VIII concentrate (Hemate P) in von Willebrand's disease 1989;56:212-217. 13. Ludlam CA. Treatment of haemophilia. Br J Haematol. 1998;101(Suppl 1):13-14. 14. Vlot AJ, Koppelman SJ, Bouma BN, et al. Factor VIII and von Willebrand factor. Thromb Haemost. 1998;79( National 15. Hemophilia Foundation (NHF), Medical and Scientific Advisory Council (MASAC). MASAC recomm concerning the treatment of hemophilia and other bleeding disorders. MASAC Recommendation #141. New NHF; revised March 2003. Available at: http://www.hemophilia.org/programs/masac/masac/masac141.htm. May 7, 2003. 16. National Hemophilia Foundation (NHF), Medical and Scientific Advisory Council (MASAC). MASAC recomm regarding the use of recombinant clotting factor replacement therapies. MASAC Recommendation #106. Ne NHF; November 12, 2000. Available at: http://www.hemophilia.org/programs/masac/masac/masac106.htm. May 7, 2003. U.S. Department of Health and Human Services (DHHS), Office of HIV/AIDS Policy, Advisory Committee on Safety and 17. Availability. Blood safety resolution - April 1998. Washington, DC: DHHS; May 8, 1998. Availabl http://www.hhs.gov/bloodsafety/resolutions/resapr98.html. Accessed March 4, 2002. Association of Hemophilia Clinic Directors of Canada (AHCDC). Hemophilia and von Willebrand's disease: 2 18. Management. Edition 2, Update 2. Toronto, ON: AHCDC; July 7, 1999. Available at: http://ahcdc.medical.org/Cpg2.html. Accessed March 4, 2002. http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014 Clotting Factors Page 25 of31 19. Anthony D, Milne R. On-demand recombinant factor VIII for people with haemophilia A. DEC Report No. 71 Southampton, UK: Wessex Institute for Health Research and Development, University of Southampton; 199 at: http://www.doh.gov.uk/research/swro/rd/publicat/dec/dec71.htm. Accessed March 4, 2002. 20. National Hemophilia Foundation (NHF), Medical and Scientific Advisory Council (MASAC). MASAC recomm concerning prophylaxis (prophylactic administration of clotting factor concentrate to prevent bleeding). MASA Recommendation #117. New York, NY: NHF; June 9, 2001. Available at: http://www.hemophilia.org/programs/masac/masac/masac117.htm. Accessed May 7, 2003. 21. Shapiro AD, Donfield SM, Lynn HS, et al. Defining the impact of hemophilia: The Academic Achievement in with 22. 23. 24. Hemophilia Study. Pediatrics. 2001;108(6):E105. van den Berg HM, Fischer K, Mauser-Bunschoten EP, et al. Long-term outcome of individualized prophylac of children with severe haemophilia. Br J Haematol. 2001;112(3):561-565. Fischer K, Astermark J, van der Bom JG, et al. Prophylactic treatment for severe haemophilia: Comparison intermediate-dose to a high-dose regimen. Haemophilia. 2002;8(6):753-760. Santagostino E, Gringeri A, Mannucci PM. State of care for hemophilia in pediatric patients. Paediatr Drugs (3):149-157. 25. Fischer K, van der Bom JG, Mauser-Bunschoten EP, et al. The effects of postponing prophylactic treatment term 26. outcome in patients with severe hemophilia. Blood. 2002;99(7):2337-2341. Inhibitor Subcommittee of the Association of Hemophilia Clinic Directors of Canada (AHCDC). Suggestions management of hemophiliacs and nonhemophiliacs with factor VIII inhibitors. 3rd ed. Association of Hemop Directors of Canada. Toronto, ON; AHCDC; 1999. Available at: http://www.ahcdc.ca/Suggestions.htm. Acce October 28, 2003. 27. Stachnik J. lnhibitor development in hemophilia A. Continuing Medical Education, Module 2. Deerfield, IL: B Hyland Immuno and Illinois Council of Health System Pharmacists; updated 2003. Available at: http://www.hemophiliagalaxy.com/2_PROFESSIONALS/MEDICAL_EDUCATION/intro.html. Accessed Octo 28. 29. 30. 31. 2003. Kroner B. Comparison of the International Immune Tolerance Registry and the North American Immune Tol Registry. Vox Sang. 1999;77(suppl 1):3337. DiMichele D, Kroner B. ISTH Factor VIII/IX Subcommittee Members. Analysis of the North American Immun Registry (NAITR) 1993-1997: Current practice implications. Vox Sang. 1999;77(suppl 1):3132. Baxter Healthcare Corporation. Advate Antihemophilic Factor (Recombinant), Plasma/Albumin-Free Metho PFM). Prescribing Information. Westlake Village, CA: Baxter; July 2003. U.S. Food and Drug Administration (FDA), Center for Biologics Evaluation and Research. Antihemophilic fa (recombinant), plasma/albumin free method. Tradename: Advate. Product Approval Information - Licensing Rockville, MD: FDA; updated August 22, 2003. Available at: http://www.fda.gov/cber/products/antibax07250 Accessed November 7, 2003. 32. Adis Data Information BV. Factor VIII - Baxter: rAHF-PFM, recombinant anti-haemophilic factor - protein-free recombinant factor VIII - protein-free. Adis R&D Profile. Drugs R D. 2003;4(6):366-368. 33. Farrugia A. Evolving perspectives in product safety for haemophilia. Haemophilia. 2002;8(3):236-243. Schlesinger KW, 34. Ragni MV. Safety of the new generation recombinant factor concentrates. Expert Opin Dru 35. 36. 37. 38. 39. 2002;1(3):213-223. Suiter TM. First and next generation native rFVIII in the treatment of hemophilia A. What has been achieved patients be switched safety. Semin Thromb Hemost. 2002;28(3):277-284. Lusher JM. First and second generation recombinant Factor VII concentrates in previously untreated patien Recovery, safety, efficacy, and inhibitor development. Semin Thromb Hemost. 2002;38(3):273-276. Brinkhous K, Sandberg H, Widlund L. et al. Preclinical pharmacology of albumin-free B-domain deleted reco factor VIII. Semin Thromb Hemost. 2002;28(3):269-272. Rothschild C, Scharrer I, Brackmann HH, et al. European data of a clinical trial with a sucrose formulated re factor VIII in previously treated haemophilia A patients. Haemophilia. 2002;8 Suppl 2:10-14. Giangrande PL, Bayer Study Group. Safety and efficacy of Kogenate® Bayer in previously untreated patien and minimally treated patients (MTPs). Haemophilia. 2002;8 Suppl 2:19-22. http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014 Clotting Factors Page 26 of31 40. Lillicrap D, Poon MC, Walker I, et al., and the Association of Hemophilia Clinic Directors of Canada. Efficacy of the factor VIII/von Willebrand factor concentrate, haemate-P/humate-P: ristocetin cofactor unit dosing in von Willebrand disease. Thromb Haemost. 2002;87(2):224-230. 41. Dobrkovska A, Krzensk U, Chediak JR. Pharmacokinetics, efficacy and safety of Humate-P in von Willebran Haemophilia. 1998;4 Suppl 3:33-39. 42. Association of Hemophilia Clinic Directors of Canada. Hemophilia and von Willebrand's disease: 2. Manage CMAJ. 1995;153(2):147-157. 43. Astermark J. Treatment of the bleeding inhibitor patient. Semin Thromb Hemost. 2003;29(1):77-86. 44. Lloyd Jones M, Wight J, Paisley S, Knight C. Control of bleeding in patients with haemophilia A with inhibito systematic review. Haemophilia. 2003;9(4):464-520. 45. Giangrande PL. Treatment of patients with haemophilia and inhibitory antibodies. Indian J Pediatr. 2003;70( National 46. Horizon Scanning Centre (NHSC). Recombinant factor VIIa in haemostatis - horizon scanning revi Birmingham, UK: 47. 48. NHSC; 2002. Berntorp E, Astermark J, Bjorkman S, et al. Consensus perspectives on prophylactic therapy for haemophili statement. Haemophilia. 2003;9(Suppl 1):1-4. Butler RB, McClure W, Wulff K. Practice patterns in haemophilia A therapy - a survey of treatment centres i States. Haemophilia. 2003;9(5):549-554. 49. d'Oiron R, Menart C, Trzeciak MC, et al. Use of recombinant factor VIIa in 3 patients with inherited type I Gl 50. 51. thrombasthenia undergoing invasive procedures. Thromb Haemost. 2000;83(5):644-647. Poon MC, d'Oiron R, Hann I, et al. Use of recombinant factor VIIa (NovoSeven) in patients with Glanzmann thrombasthenia. Semin Hematol. 2001;38(4 Suppl 12):21-25. Erhardtsen E. To general haemostasis--the evidence-based route. Pathophysiol Haemost Thromb. 2002;32 52. 52. Lloyd JV, Joist JH. Recombinant factor VIIa: A universal hemostatic agent? Curr Hematol Rep. 2002;1(1):1 53. Devecioglu O, Unuvar A, Anak S, et al. Pyelolithotomy in a patient with Glanzmann thrombasthenia and antiglycoprotein IIb/IIIa antibodies: The shortest possible duration of treatment with recombinant activated fa 54. platelet transfusions. Turk J Pediatr. 2003;45(1):64-66. Almeida AM, Khair K, Hann I, Liesner R. The use of recombinant factor VIIa in children with inherited platel disorders. 55. Br J Haematol. 2003;121(3):477-481. Caglar K, Cetinkaya A, Aytac S, et al. Use of recombinant factor VIIa for bleeding in children with Glanzman 56. thrombasthenia. Pediatr Hematol Oncol. 2003;20(6):435-438. Lloyd Jones M, Wight J, Paisley S, Knight C. Control of bleeding in patients with haemophilia A with inhibito 57. systematic review. Haemophilia. 2003;9(4):464-520. 58. Escolar G. New insights into the management of bleeding disorders. Drug News Perspect. 2004;17(4):285- Hind D, Lloyd-Jones M, Makris M, Paisley S. Recombinant Factor VIIa concentrate versus plasma derived c for the treatment of acute bleeding episodes in people with Haemophilia A and inhibitors. Cochrane Databas Rev. 2004;(2):CD004449. 59. Mayer SA, Brun NC, Broderick J, et al. Safety and feasibility of recombinant factor VIIa for acute intracerebr hemorrhage. Stroke. 2005;36(1):74-79. 60. Broderick JP. Advances in the treatment of hemorrhagic stroke: A possible new treatment. Cleve Clin J Med (4):341-344. 61. Levi M, Peters M, Buller HR.. Efficacy and safety of recombinant factor VIIa for treatment of severe bleedin systematic review. Crit Care Med. 2005;33(4):883-890. 62. Lam MS, Sims-McCallum RP. Recombinant factor VIIa in the treatment of non-hemophiliac bleeding. Ann Pharmacother. 2005;39(5):885-891. 63. Lodge JP, Jonas S, Oussoultzoglou E, et al. Recombinant coagulation factor VIIa in major liver resection: A randomized, placebo-controlled, double-blind clinical trial. Anesthesiology. 2005;102(2):269-275. 64. O'Connell KA, Wood JJ, Wise RP, et al. Thromboembolic adverse events after use of recombinant human c factor VIIa. JAMA. 2006;295(3):293-298. http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014 Clotting Factors Page 27 of31 65. National Horizon Scanning Centre (NHSC). Recombinant factor VIIa (NovoSeven) for life-threatening surgic traumatic bleeding in non-haemophiliacs - horizon scanning review. Birmingham, UK: National Horizon Sca Centre (NHSC); 2003:1-4. 66. National Horizon Scanning Centre (NHSC). Recombinant factor VIIa (NovoSeven) in intracerebral haemorrh horizon scanning review. Birmingham, UK: National Horizon Scanning Centre (NHSC); 2004:1-5. 67. National Horizon Scanning Centre (NHSC). Recombinant factor VIIa (NovoSeven) for prevention and treatm bleeding in patients with underlying disease - horizon scanning review. Birmingham, UK: National Horizon S Centre (NHSC); 2003:1-7. 68. European Medicines Agency (EMEA). Novoseven INN:Eptacog alfa (activated). Scientific Discussion. Euro Assessment Report. London, UK: EMEA; 2005. Available at: 69. http://www.emea.eu.int/humandocs/PDFs/EPAR/Novoseven/072995en6.pdf. Accessed February 6, 2006. Danish Centre for Evaluation and Health Technology Assessment (DACEHTA). NovoSeven for massive, uncontrollable, lifethreatening haemorrhage in non-haemophiliacs. Early Warning on New Health Technolo Copenhagen, Denmark: DACEHTA; 2003;2(1). Available at: http://www.cemtv.dk/tidligVarsling/varsler/pdf/NovoSeven.pdf. Accessed March 70. 12, 2007. BlueCross BlueShield Association (BCBSA), Technology Evaluation Center (TEC). Special report: Recombi activated factor VII for uncontrolled bleeding in non-hemophiliac patients. TEC Assessment Program. Chica BCBSA; October 2006;21(10). Available at: http://www.bcbs.com/betterknowledge/tec/vols/21/21_10.html. A March 12, 2007. 71. Stobart K, Iorio A, Wu JK. Clotting factor concentrates given to prevent bleeding and bleeding-related comp people with hemophilia A or B. Cochrane Database Syst Rev. 2006;(2):CD003429. 72. Adelaide Health Technology Assessment (AHTA) on behalf of the National Blood Authority. Clinical practice and national tolerisation protocols for the use of recombinant and plasma derived factor VIII and factor IX pr Adelaide, SA: Adelaide Health Technology Assessment (AHTA) on behalf of the National Blood Authority; 2 73. Selin S, Tejani A. Recombinant activated factor VII for bleeding in patients without inherited bleeding disord in Emerging Health Technologies Issue 82. Ottawa, ON: Canadian Coordinating Office for Health Technolog Assessment (CCOHTA); 2006. 74. You H, Al-Shahi R. Haemostatic drug therapies for acute primary intracerebral haemorrhage. Cochrane Dat Rev. 2006(3):CD005951. 75. NHS Quality Improvement Scotland (NHS QIS). Evidence note 17: The use of recombinant factor VIIa (rFVI people with haemophilia who develop inhibitors. Glasgow: NHS Quality Improvement Scotland (NHS QIS); Marti-Carvajal AJ, 76. Salanti G, Marti-Carvajal PI. Human recombinant activated factor VII for upper gastrointe bleeding in patients with liver diseases. Cochrane Database Syst Rev. 2007;(1):CD004887. 77. Stanworth SJ, Birchall J, Doree CJ, Hyde C. Recombinant factor VIIa for the prevention and treatment of ble patients without haemophilia. Cochrane Database Syst Rev. 2007;(2):CD005011. 78. Carter NJ, Scott LJ. Human plasma von Willebrand factor/factor VIII complex (Haemate P/Humate-P): In vo Willebrand disease and haemophilia A. Drugs. 2007;67(10):1513-1519. 79. U.S. Food and Drug Administration (FDA). FDA approves product to treat common bleeding disorder. FDA Rockville, MD: FDA; April 27, 2007. Available at: http://www.fda.gov/bbs/topics/NEWS/2007/NEW01619.htm January 24, 2008. Sattin JA, Zivin JA. Emerging therapies for acute ischemic stroke. Am J Ther. 2007;14(3):291-298. 80. Bartal C, Freedman J, Bowman K, Cusimano M. Coagulopathic patients with traumatic intracranial bleeding the role of 81. recombinant factor VIIa. J Trauma. 2007;63(4):725-732. 82. Franchini M, Manzato F, Salvagno GL, Lippi G. Potential role of recombinant activated factor VII for the trea severe bleeding associated with disseminated intravascular coagulation: A systematic review. Blood Coagu Fibrinolysis. 2007;18(7):589-593. 83. Girona E, Borrás-Blasco J, Conesa-García V, et al. Successful treatment of severe gastrointestinal bleeding to Crohn disease with recombinant factor VIIa. South Med J. 2007;100(6):601-604. http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014 Clotting Factors Page 28 of31 84. Narayan RK, Maas AI, Marshall LF, et al; rFVIIa Traumatic ICH Study Group. Recombinant factor VIIA in tra intracerebral hemorrhage: Results of a dose-escalation clinical trial. Neurosurgery. 2008;62(4):776-786; dis -788. 85. Mayer SA, Brun NC, Begtrup K, et al; FAST Trial Investigators. Efficacy and safety of recombinant activated for acute intracerebral hemorrhage. N Engl J Med. 2008;358(20):2127-2137. 86. Bosch J, Thabut D, Albillos A, et al; International Study Group on rFVIIa in UGI Hemorrhage. Recombinant for variceal bleeding in patients with advanced cirrhosis: A randomized, controlled trial. Hepatology. 2008;47 1614. 87. Baxter Healthcare Corporation. Feiba VH. Anti-inhibitor coagulant complex. vapor heated. Prescribing Infor Westlake Village, CA: Baxter; April 2005. Available at: http://www.thereforyou.com/pdf/feiba-pi.pdf. Accesse 1, 2010. 88. Joshi AV, Stephens JM, Munro V, et al. Pharmacoeconomic analysis of recombinant factor VIIa versus APC treatment of minor-to-moderate bleeds in hemophilia patients with inhibitors. Curr Med Res Opin. 2006;22(1 89. Gomperts ED, Astermark J, Gringeri A, Teitel J. From theory to practice: Applying current clinical knowledg treatment 90. 91. 92. strategies to the care of hemophilia A patients with inhibitors. Blood Rev. 2008;22 Suppl 1:S1-S11 Steen Carlsson K, Astermark J, Donfield S, Berntorp E. Cost and outcome: Comparisons of two alternative agents for persons with haemophilia A complicated by an inhibitor. Thromb Haemost. 2008;99(6):1060-106 Tiede A, Tait RC, Shaffer DW, et al. Antithrombin alfa in hereditary antithrombin deficient patients: A phase prophylactic intravenous administration in high risk situations. Thromb Haemost. 2008;99(3):616-622. Afshari A, Wetterslev J, Brok J, Møller AM. Antithrombin III for critically ill patients. Cochrane Database Syst (3):CD005370. 93. Talecris Biotherapeutics, Inc. Thrombate III. Antithrombin III (human). Prescribing Information. Research Tri NC: Talecris; May 2009. Available at: http://www.talecris-pi.info/inserts/thrombate.pdf. Accessed February 1 94. Federici AB. The safety of plasma-derived von Willebrand/factor VIII concentrates in the management of inh Willebrand disease. Expert Opin Drug Saf. 2009;8(2):203-210. 95. Youjin S, Jun Y. The treatment of hemophilia A: From protein replacement to AAV-mediated gene therapy. Lett. 2009;31(3):321-328. 96. Rogers GM. Role of antithrombin concentrate in treatment of hereditary antithrombin deficiency. An update. Haemost. 2009;101(5):806-812. 97. Berntorp E. Differential response to bypassing agents complicates treatment in patients with haemophilia an Haemophilia. 2009;15(1):3-10. Treur MJ, McCracken F, Heeg B, et al. Efficacy of recombinant activated factor VII vs. activated prothrombi 98. concentrate for patients suffering from haemophilia complicated with inhibitors: A Bayesian meta-regression Haemophilia. 2009;15(2):420-436. 99. Raobaikady R, Redman J, Ball JA, et al. Use of activated recombinant coagulation factor VII in patients und reconstruction surgery for traumatic fracture of pelvis or pelvis and acetabulum: A double-blind, randomized controlled trial. Br J Anaesth. 2005;94(5):586-591. 100. Lovejoy AE, Reynolds TC, Visich JE, et al. Safety and pharmacokinetics of recombinant factor XIII-A2 admi patients with congenital factor XIII deficiency. Blood. 2006;108(1):57-62. 101. Hardy JF, Bélisle S, Van der Linden P. Efficacy and safety of activated recombinant factor VII in cardiac sur patients. Curr Opin Anaesthesiol. 2009;22(1):95-99. 102. Levi M, Levy JH, Andersen HF, Truloff D. Safety of recombinant activated factor VII in randomized clinical tr J Med. 2010;363(19):1791-1800. 103. Aledort LM. Off-label use of recombinant activated factor VII -- safe or not safe? N Engl J Med. 2010;363(19 1854. 104. Murphy G, Tsakonas E, Ndegwa S, et al. Recombinant activated factor VII for prevention of bleeding unrela hemophilia: Clinical and economic systematic review [Technology report number 124]. Canadian Agency fo Technologies in Health (CADTH), February 2010. Available at: http://www.cadth.ca/media/pdf/457B_Executive_Summary_e.pdf. accessed December 30, 2010. http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014 Clotting Factors Page 29 of31 105. Yuan ZH, Jiang JK, Huang WD, et al. A meta-analysis of the efficacy and safety of recombinant activated fa patients with acute intracerebral hemorrhage without hemophilia. J Clin Neurosci. 2010;17(6):685-693. Mannucci PM, 106. Franchini M. Mechanism of hemostasis defects and management of bleeding in patients with coronary syndromes. Eur J Intern Med. 2010;21(4):254-259. 107. Perel P, Roberts I, Shakur H, et al. Haemostatic drugs for traumatic brain injury. Cochrane Database Syst R (1):CD007877. 108. Valentino LA. Assessing the benefits of FEIBA prophylaxis in haemophilia patients with inhibitors. Haemoph 2010;16(2):263-271. 109. Mannucci PM, Duga S, Peyvandi F. Rare (recessively inherited) coagulation disorders. September 2010. Up Waltham, MA. 110. U.S. Food and Drug Administration. FDA approves product to prevent bleeding in people with rare genetic d News. Rockville, MD: FDA; February 17, 2011. Available at: 111. 112. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm243856.htm. Accessed February 18, Logan AC, Yank V, Stafford RS. Off-label use of recombinant factor VIIa in U.S. hospitals: Analysis of hospi Ann Intern Med. 2011;154(8):516-522. Yank V, Tuohy CV, Logan AC, et al. Systematic review: Benefits and harms of in-hospital use of recombina VIIa for off-label indications. Ann Intern Med. 2011;154(8):529-540. 113. Avorn J, Kesselheim A. A hemorrhage of off-label use. Ann Intern Med. 2011;154(8):566-567. 114. Koncar IB, Davidovic LB, Savic N, et al. Role of recombinant factor VIIa in the treatment of intractable bleed vascular 115. 116. surgery. J Vasc Surg. 2011;53(4):1032-1037. Witmer CM, Huang YS, Lynch K, et al. Off-label recombinant factor VIIa use and thrombosis in children: A m cohort study. J Pediatr. 2011;158(5):820-825. Lin Y, Stanworth S, Birchall J, et al. Recombinant factor VIIa for the prevention and treatment of bleeding in without 117. haemophilia. Cochrane Database Syst Rev. 2011;(2):CD005011. Highlights of Prescribing Information. Wilate, von Willebrand factor/coagulation factor VIII complex (human) at: http://www.fda.gov/downloads/BiologicsBloodVaccines/BloodBloodProducts/ApprovedProducts/LicensedPro FractionatedPlasmaProducts/UCM193127.pdf. Accessed November 7, 2012. 118. Hoots WK, Shapiro AD. Factor VIII and factor IX inhibitors in patients with hemophilia. Last reviewed Octobe UpToDate Inc. Waltham, MA. 119. Hoots WK, Shapiro AD. Treatment of hemophilia. Last reviewed October 2012b. UpToDate Inc. Waltham, M BeneFIX 120. [Coagulation Factor IX (Recombinant)]. November 2011. Wyeth Pharmaceuticals Inc.: Philadelphia Available at: http://labeling.pfizer.com/showlabeling.aspx?id=492. Accessed August 12, 2013. 121. Franchini M, Frattini F, Crestani S, et al. Treatment of hemophilia B: Focus on recombinant factor IX. Biolog 2013;7:33-38. 122. Food and Drug Administration. FDA approves first recombinant coagulation factor IX that is specifically indi routine use in preventing bleeding episodes (prophylaxis). June 27, 2013. FDA: Silver Spring, MD. Available http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm358918.htm. Accessed August 12, 2 123. Hoots WK, Shapiro AD. Treatment of hemophilia. Last reviewed July 2013. UpToDate Inc., Waltham, MA. Windyga J, 124. Lissitchkov T, Stasyshyn O, et al. Pharmacokinetics, efficacy and safety of BAX326, a novel rec factor IX: a prospective, controlled, multicentre phase I/III study in previously treated patients with severe (F <1%) or moderately severe (FIX level ≤2%) haemophilia B. Haemophilia. 2014;20(1):15-24. 125. National Institute for Health and Clinical Excellence (NICE). Acute upper gastrointestinal bleeding: Managem London (UK): NICE; June 2012. (Clinical guideline; no. 141). Available at: http://www.guideline.gov/content. 126. 127. id=37563&search=prothrombin+complex+concentrate+. Accessed September 27, 2013. Hickey M, Gatien M, Taljaard M, et al. Outcomes of urgent warfarin reversal with frozen plasma versus proth complex concentrate in the emergency department. Circulation. 2013;128(4):360-364. Hanke AA, Joch C, Gorlinger K. Long-term safety and efficacy of a pasteurized nanofiltrated prothrombin co concentrate (Beriplex P/N): A pharmacovigilance study. Br J Anaesth. 2013;110(5):764-772. http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014 Page 30 of31 Clotting Factors 128. Sarode R, Milling TJ Jr, Refaai MA, et al. Efficacy and safety of a 4-factor prothrombin complex concentrate on vitamin K antagonists presenting with major bleeding: A randomized, plasma-controlled, phase IIIb study Circulation. 2013;128(11):1234-1243. 129. Food and Drug Administration. FDA approves Kcentra for the urgent reversal of anticoagulation in adults wit bleeding. April 29, 2013. FDA: Silver Spring, MD. Available at: http://www.fda.gov/NewsEvents/Newsroom/Pressannouncements/ucm350026.htm. Accessed September 2 130. No authors listed. Kcentra (Prothrombin Complex Concentrate (Human)) prescribing Information. CSL Behr Kankakee, IL. 2013. Available at: http://labeling.cslbehring.com/PI/US/Kcentra/EN/Kcentra-Prescribing-Infor Accessed September 27, 2013. 131. Ferreira J, DeLosSantos M. The clinical use of prothrombin complex concentrate. J Emerg Med. 2013;44(6) Dewhirst E. 132. Towards evidence-based emergency medicine: Best BETs from the Manchester Royal Infirmar recombinant factor VIIa beneficial in the management of acute spontaneous intracerebral hemorrhage? Em 133. 134. 135. 2013;30(4):340-341. Awad NI, Cocchio C. Activated prothrombin complex concentrates for the reversal of anticoagulant-associat coagulopathy. P T. 2013;38(11):696-701. Stewart WS, Pettit H. Experiences with an activated 4-factor prothrombin complex concentrate (FEIBA) for warfarinrelated bleeding. Am J Emerg Med. 2013;31(8):1251-1254. Cabral KP, Fraser GL, Duprey J, et al. Prothrombin complex concentrates to reverse warfarin-induced coag patients 136. with intracranial bleeding. Clin Neurol Neurosurg. 2013;115(6):770-774. Song HK, Tibayan FA, Kahl EA, et al. Safety and efficacy of prothrombin complex concentrates for the treat 137. coagulopathy after cardiac surgery. J Thorac Cardiovasc Surg. 2013 Dec 20. [Epub ahead of print] U.S. Food and Drug Administration (FDA). FDA approves Tretten to treat rare genetic clotting disorder. FDA Release. 138. Silver Spring, MD: FDA; December 23, 2013. Novo Nordisk, Inc. Tretten, Coagulation Factor XIII A-Subunit (Recombinant) for Intravenous Use. Lyophiliz for 139. Solution for Injection. Prescribing Information. Plainsboro, NJ: Novo Nordisk; December 2013. U.S. Food and Drug Administration (FDA). FDA approves first long-acting recombinant coagulation Factor IX 140. concentrate for patients with Hemophilia B. FDA News Release. Silver Spring, MD: FDA; March 28, 2013. Biogen Idec Inc. Alprolix™ [Coagulation Factor IX (Recombinant), Fc Fusion Protein], Lyophilized Powder f For Intravenous Injection. 141. Prescribing Information. 43297-02 . Cambridge, MA: Biogen Idec; revised March 2 Powell JS, Pasi KJ, Ragni MV, et al.; B-LONG Investigators. Phase 3 study of recombinant factor IX Fc fusi hemophilia B. 142. N Engl J Med. 2013;369(24):2313-2323. Biogen Idec Inc. FDA approves Biogen Idec’s Elocate™, first hemophilia A therapy to extend the interval be 143. prophylactic infusions, for both adults and children. Press Release. Cambridge, MA: Biogen Idec; June 6, 2 Sommer JM, Moore N, McGuffie-Valentine B, et al. Comparative ï¬ eld study evaluating the activity of reco factor VIII Fc fusion protein in plasma samples at clinical haemostasis laboratories. Haemophilia. 2014;20: 144. Biogen Idec Inc.. Eloctate™ [antihemophilic factor (recombinant), Fc fusion protein] lyophilized powder for s intravenous injection. Prescribing Information. 44279-01. Cambridge, MA: Biogen Idec; revised June 2014. http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014 Clotting Factors Page 31 of31 Copyright Aetna Inc. All rights reserved. Clinical Policy Bulletins are developed by Aetna to assist in administering plan benefits and constitute neithe coverage nor medical advice. This Clinical Policy Bulletin contains only a partial,general description of plan or program benefits and does not constit Aetna does not provide health care services and, therefore, cannot guarantee any results or outcomes. Participating providers are independent contr practice and are neither employees nor agents of Aetna or its affiliates. Treating providers are solely responsible for medical advice and treatment of Clinical Policy Bulletin may be updated and therefore is subject to change. CPT only copyright 2008 American Medical Association. All Rights Reserved. http://qawww.aetna.com/cpb/medical/data/100_199/0131_draft.html 10/22/2014