
2 a - Atelier de BioInformatique
... • Cliques of “symbols” define similarity, e.g : - different overlapping sets of amino acids clustered by their properties (e.g. hydrophobic, hydrophilic, small, large, polar, charged, etc…) - different overlapping sets of amino acids clustered by setting a threshold value on their score in a similar ...
... • Cliques of “symbols” define similarity, e.g : - different overlapping sets of amino acids clustered by their properties (e.g. hydrophobic, hydrophilic, small, large, polar, charged, etc…) - different overlapping sets of amino acids clustered by setting a threshold value on their score in a similar ...

Srivastava, Sanjay: Analysis of Methods for Predicting Protein Fold and Remote Homologue Recognition
... protein structure. The most widely used methods involve either using pairwise searches or using structural information. The pairwise search method uses a single sequence of the unknown protein is scanned against each sequence in a database using programs such as BLAST, FASTA or any dynamic programmi ...
... protein structure. The most widely used methods involve either using pairwise searches or using structural information. The pairwise search method uses a single sequence of the unknown protein is scanned against each sequence in a database using programs such as BLAST, FASTA or any dynamic programmi ...

Slides
... identify a given individual. Similarly, a motif can be used to assign a newly sequenced protein to a specific family of proteins and thus to formulate hypotheses about its function. ...
... identify a given individual. Similarly, a motif can be used to assign a newly sequenced protein to a specific family of proteins and thus to formulate hypotheses about its function. ...

Explorging Food Science Unit 2 Glossaries
... Sanitation as applied in food safety is the second step after thorough cleansing and rinsing of a surface or utensil that comes into contact with food. The final step of sanitation uses a mild solution of water and bleach. This last step destroys most pathogenic (disease causing) bacteria. ...
... Sanitation as applied in food safety is the second step after thorough cleansing and rinsing of a surface or utensil that comes into contact with food. The final step of sanitation uses a mild solution of water and bleach. This last step destroys most pathogenic (disease causing) bacteria. ...

Amino Acid Starter Kit©
... and keeping all of the polar white tacks on the surface of the protein. After everyone has folded their Toober, the teacher can point out: ...
... and keeping all of the polar white tacks on the surface of the protein. After everyone has folded their Toober, the teacher can point out: ...

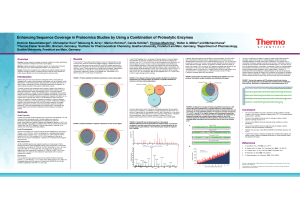
Enhancing Sequence Coverage in Proteomics
... common enzyme of choice for proteomics experiments. Digestion with trypsin (or any single enzyme in general) often results in the identification of large numbers of proteins, but sequence coverage is frequently incomplete. If maximum sequence coverage is desired (e.g. when studying changes in protei ...
... common enzyme of choice for proteomics experiments. Digestion with trypsin (or any single enzyme in general) often results in the identification of large numbers of proteins, but sequence coverage is frequently incomplete. If maximum sequence coverage is desired (e.g. when studying changes in protei ...

Anti-UBR1 Antibody
... heart. In mouse embryo, UBR1 is primarily expressed in the branchial arches and in the tail and limb buds. UBR1 is located on mouse chromosome 2 and on human chromosome 15 in the syntenic region. The UBR1-selective antibodies were generated against unique N-terminal peptides characteristics of the p ...
... heart. In mouse embryo, UBR1 is primarily expressed in the branchial arches and in the tail and limb buds. UBR1 is located on mouse chromosome 2 and on human chromosome 15 in the syntenic region. The UBR1-selective antibodies were generated against unique N-terminal peptides characteristics of the p ...

The influence of membrane lipid structure on plasma
... gene products, and its network link means simple physical binding rather than in-depth knowledge of biological process. In this paper, we suggest a protein functional flow model which is a directed network based on a protein functions’ relation of signaling transduction pathway. The vertex of the su ...
... gene products, and its network link means simple physical binding rather than in-depth knowledge of biological process. In this paper, we suggest a protein functional flow model which is a directed network based on a protein functions’ relation of signaling transduction pathway. The vertex of the su ...

Amino Acids
... • First step in determining 1º structure of a polyp. is to identify and quantitate its aa’s. • A purified sample of a polyp. to be analyzed is hydrolyzed by strong acid at 110 ºC for 24 h. – Treatment cleaves peptide bonds and releases free aa’s separated by cation-exchange chromatography. – The a ...
... • First step in determining 1º structure of a polyp. is to identify and quantitate its aa’s. • A purified sample of a polyp. to be analyzed is hydrolyzed by strong acid at 110 ºC for 24 h. – Treatment cleaves peptide bonds and releases free aa’s separated by cation-exchange chromatography. – The a ...

Confocal Fluorescence Microscopy
... A confocal fluorescence microscope is a serial rather than parallel imageacquisition device: the object is illuminated point by point and the generated fluorescence, imaged onto the detection pinhole, is measured sequentially for each illuminated point. In such an instrument, the image acquisition is ...
... A confocal fluorescence microscope is a serial rather than parallel imageacquisition device: the object is illuminated point by point and the generated fluorescence, imaged onto the detection pinhole, is measured sequentially for each illuminated point. In such an instrument, the image acquisition is ...

MB207_7 - MB207Jan2010
... – Empty tRNA is ejected and the peptidyl-tRNA is shifted from the A site to the P site – New aminoacyl-tRNA is allowed to enter within the A site – Translocation is catalyzed by the elongation factor EF-G. The G indicates that this factor uses the energy gained from the hydrolysis of GTP for translo ...
... – Empty tRNA is ejected and the peptidyl-tRNA is shifted from the A site to the P site – New aminoacyl-tRNA is allowed to enter within the A site – Translocation is catalyzed by the elongation factor EF-G. The G indicates that this factor uses the energy gained from the hydrolysis of GTP for translo ...

Facts and Fallacies
... • Better conduct de novo on all spectra. – De novo not slow, and computing is cheap. – De novo provides independent validation for DB result. # consensus AA (de novo vs. DB search) ...
... • Better conduct de novo on all spectra. – De novo not slow, and computing is cheap. – De novo provides independent validation for DB result. # consensus AA (de novo vs. DB search) ...

Hydrogen Bonds, Hydrophobicity Forces and the Character of the
... computational limitations and uncertainties about the relevant forces, which makes model building a delicate and highly relevant task. Most current models use one or both of two quite drastic approximations, the lattice and Gō [1] approximations, where the latter amounts to ignoring interactions th ...
... computational limitations and uncertainties about the relevant forces, which makes model building a delicate and highly relevant task. Most current models use one or both of two quite drastic approximations, the lattice and Gō [1] approximations, where the latter amounts to ignoring interactions th ...

Laemmli Buffer Recipe Preparation
... Bromophenol blue also functions to make it easier to see the sample during loading of the gel wells with protein sample.Glycerol in the Laemmli buffer increases the density of the sample so that it will fall to the bottom of the well, minimizing puffing or loss of protein sample in the buffer, and l ...
... Bromophenol blue also functions to make it easier to see the sample during loading of the gel wells with protein sample.Glycerol in the Laemmli buffer increases the density of the sample so that it will fall to the bottom of the well, minimizing puffing or loss of protein sample in the buffer, and l ...

TD11 Identification of in vivo substrates of GroEL Nature 1999, 402
... Figure 1c shows after anti-GroEL IP now there are only 250-300 spots- 10% compared to control (indicates proteins that interact with GroEL), notice the predominance of larger proteins Figures 1e and f show how pI (isoelectric point) and MW compare to total proteins -pI distribution is about the same ...
... Figure 1c shows after anti-GroEL IP now there are only 250-300 spots- 10% compared to control (indicates proteins that interact with GroEL), notice the predominance of larger proteins Figures 1e and f show how pI (isoelectric point) and MW compare to total proteins -pI distribution is about the same ...

Question 1 - University of Missouri
... Conformations New NMR techniques can gather local conformations and long-range interactions even under strongly denaturing conditions to obtain plausible all-atom models of the unfolded state at increasing accuracy. ...
... Conformations New NMR techniques can gather local conformations and long-range interactions even under strongly denaturing conditions to obtain plausible all-atom models of the unfolded state at increasing accuracy. ...

Gene Section PTPN14 (protein tyrosine phosphatase, non receptor type 14) -
... TGF-β: PTPN14 promotes epithelial-mesenchymal transition (EMT) via increased TGF-beta production in MDCK epithelial cells (Wyatt et al., 2007) Lymphangiogenesis: PTPN14 forms a complex with ...
... TGF-β: PTPN14 promotes epithelial-mesenchymal transition (EMT) via increased TGF-beta production in MDCK epithelial cells (Wyatt et al., 2007) Lymphangiogenesis: PTPN14 forms a complex with ...

Document
... have identified thousands of novel protein-protein interactions good: pull down multiprotein complexes, providing a more realistic picture of interactions possible to see interactions that are dependent upon PTMs can do this type of analysis in relevant organism/cell/tissue not so good: lots of non- ...
... have identified thousands of novel protein-protein interactions good: pull down multiprotein complexes, providing a more realistic picture of interactions possible to see interactions that are dependent upon PTMs can do this type of analysis in relevant organism/cell/tissue not so good: lots of non- ...



doc - DePaul University
... the overall stability of the protein while the non-core amino acids play a significant role in determining the tertiary structure. The results were encouraging, demonstrating that it was possible to repack the core alone with novel sequences. Unfortunately, the potential sequences were limited by th ...
... the overall stability of the protein while the non-core amino acids play a significant role in determining the tertiary structure. The results were encouraging, demonstrating that it was possible to repack the core alone with novel sequences. Unfortunately, the potential sequences were limited by th ...

Expanded protein information at SGD: new pages and proteome browser.
... describes the specific function of the protein when it is known. These fields have been recently reviewed and rewritten using a standard, consistent format so that they accurately reflect the current state of knowledge for each gene product. The references for this information are found at the botto ...
... describes the specific function of the protein when it is known. These fields have been recently reviewed and rewritten using a standard, consistent format so that they accurately reflect the current state of knowledge for each gene product. The references for this information are found at the botto ...

1. Amino acids. Of all data abstractions in
... or due to functional requirements. Functional requirements could be tested if a mechanistic model for the function makes predictions for requiring specific residues. If the two structurally similar proteins are not related through common ancestry, the coincidence of a functional site in one protein ...
... or due to functional requirements. Functional requirements could be tested if a mechanistic model for the function makes predictions for requiring specific residues. If the two structurally similar proteins are not related through common ancestry, the coincidence of a functional site in one protein ...

Classification and Regression Tree (CART) Analysis for Deriving
... may influence diminutive changes in conformation ...
... may influence diminutive changes in conformation ...