ArnoldSpr09
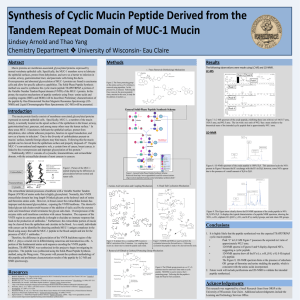
... Overexpression and abnormal glycosylation of MUC-1 proteins are found in carcinoma cells and allow for specific adhesive capabilities. The Solid-Phase Peptide Synthesis method was used to synthesize the cyclic mucin peptide TSAPDTRPAP, a portion of the Variable Number Tandem Repeat domain (VNTR) of ...
... Overexpression and abnormal glycosylation of MUC-1 proteins are found in carcinoma cells and allow for specific adhesive capabilities. The Solid-Phase Peptide Synthesis method was used to synthesize the cyclic mucin peptide TSAPDTRPAP, a portion of the Variable Number Tandem Repeat domain (VNTR) of ...

Sample exam 1
... b. Identify the Roman numeral point at the isoelectric point. Draw a predominant structure or otherwise explain your choice. 7. The protein myoglobin is found in numerous organisms, and the amino acid residue sequence of the protein from a wide variety of organisms has been determined. Recall that ...
... b. Identify the Roman numeral point at the isoelectric point. Draw a predominant structure or otherwise explain your choice. 7. The protein myoglobin is found in numerous organisms, and the amino acid residue sequence of the protein from a wide variety of organisms has been determined. Recall that ...

Spectrophotometric Determination of Total Protein
... Proteins are formed by the linkage of these amino acid groups via an amine group-carboxylic acid group covalent bond which also produces water. Hundreds to thousands of amino acids are present in proteins. There are 20 standard amino acid groups which vary only in what is called the “side chain”. Th ...
... Proteins are formed by the linkage of these amino acid groups via an amine group-carboxylic acid group covalent bond which also produces water. Hundreds to thousands of amino acids are present in proteins. There are 20 standard amino acid groups which vary only in what is called the “side chain”. Th ...

“Characterization of Proteins Interacting with Cystinosin” – Lay
... with cystinosin. This allowed us to identify proteins potentially important for cystinosin function, that were not necessarily expected from what was not known in the field of cystinosis. Indeed, we identified galectin-3, a protein known to be able to interact with sugar appended to certain proteins ...
... with cystinosin. This allowed us to identify proteins potentially important for cystinosin function, that were not necessarily expected from what was not known in the field of cystinosis. Indeed, we identified galectin-3, a protein known to be able to interact with sugar appended to certain proteins ...

slides
... given protein might have a glycine at a given position, which by itself might suggest a random coil there. However, multiple sequence alignment might reveal that helix-favoring amino acids occur at that position (and nearby positions) in 95% of homologous proteins spanning nearly a billion years of ...
... given protein might have a glycine at a given position, which by itself might suggest a random coil there. However, multiple sequence alignment might reveal that helix-favoring amino acids occur at that position (and nearby positions) in 95% of homologous proteins spanning nearly a billion years of ...

1) digest DNA inserts with restriction enzyme(s).
... Coated membrane buds that contain clathrin, adaptins, and receptors bound to their ligands pinch off to form coated vesicles. ...
... Coated membrane buds that contain clathrin, adaptins, and receptors bound to their ligands pinch off to form coated vesicles. ...

Potts Devine et al final final Supporting Information Apr 2017
... The five PDB structures were then aligned manually with one another before the C-terminal amino acid from the linker region was connected to the N-terminal leucine of the sequential I27 sub-unit. The subunits were connected using the Coot software (1), operated under a Linux operating system. ...
... The five PDB structures were then aligned manually with one another before the C-terminal amino acid from the linker region was connected to the N-terminal leucine of the sequential I27 sub-unit. The subunits were connected using the Coot software (1), operated under a Linux operating system. ...

Materials and Methods - UROP
... for 45 minutes. The supernatant was then sterile filtered through a 0.45 μm membrane. This sample was then loaded onto a nickel affinity column, washed with 40 ml of the binding buffer, and 10 ml of the wash buffer (20 mM imidazole, 500 mM NaCl, 20 mM Tris-HCl (pH 7.5)). The DNase which is bound to ...
... for 45 minutes. The supernatant was then sterile filtered through a 0.45 μm membrane. This sample was then loaded onto a nickel affinity column, washed with 40 ml of the binding buffer, and 10 ml of the wash buffer (20 mM imidazole, 500 mM NaCl, 20 mM Tris-HCl (pH 7.5)). The DNase which is bound to ...


animal science nutrition laboratory standard procedures and safety
... nitrogen and crude protein content can be calculated. An example of this calculation is shown in Table 3-4. Generally, samples to be analyzed for kjeldahl nitrogen are weighed and wrapped in filter paper and then placed in the reaction vessel. Containing the sample in filter paper is simply a method ...
... nitrogen and crude protein content can be calculated. An example of this calculation is shown in Table 3-4. Generally, samples to be analyzed for kjeldahl nitrogen are weighed and wrapped in filter paper and then placed in the reaction vessel. Containing the sample in filter paper is simply a method ...

What more do we need to know to optimize the
... – Look closely at what ingredients are in the diet • Make sure NSP enzymes are present when needed – impacts on digestibility of all other nutrients • Use of phytases will increase the proteins available for digestion – interactions with proteases? • Make sure fat levels are not to low or to high – ...
... – Look closely at what ingredients are in the diet • Make sure NSP enzymes are present when needed – impacts on digestibility of all other nutrients • Use of phytases will increase the proteins available for digestion – interactions with proteases? • Make sure fat levels are not to low or to high – ...

Proportion of animal protein Consumption
... Animal proteins are enriched in 15N in relative to plant-derived food proteins due to trophic shifts 13 C are good reflection of their plant or animal origin ...
... Animal proteins are enriched in 15N in relative to plant-derived food proteins due to trophic shifts 13 C are good reflection of their plant or animal origin ...

Analysis of High Accuracy, Quantitative Proteomics Data in the
... purposes in mind. The Global Proteome Machine (9) and PeptideAtlas (10, 11) are two of the earliest such collections, with the primary goal of providing a collection of peptide identifications. These collections can, for example, be mined for the design of multiple reaction monitoring experiments in ...
... purposes in mind. The Global Proteome Machine (9) and PeptideAtlas (10, 11) are two of the earliest such collections, with the primary goal of providing a collection of peptide identifications. These collections can, for example, be mined for the design of multiple reaction monitoring experiments in ...
MIAPE_Quant_v1.0_SILAC_MLHS
... 5.1 Quantification values at peptide and/or feature level: Actual quantification values achieved for each peptide and/or, in case of feature-based quantification, for the corresponding features (mapped back from each peptide), together with their estimated confidence. ...
... 5.1 Quantification values at peptide and/or feature level: Actual quantification values achieved for each peptide and/or, in case of feature-based quantification, for the corresponding features (mapped back from each peptide), together with their estimated confidence. ...

Deep architectures for protein contact map prediction
... in a protein sequence are spatially close to each other in the folded 3D structure. For a protein of N amino acids, the contact map is an NxN matrix C whose elements are by: ...
... in a protein sequence are spatially close to each other in the folded 3D structure. For a protein of N amino acids, the contact map is an NxN matrix C whose elements are by: ...

Screening for novel snake venom toxins using protein chemistry and molecular biology.
... We have been involved in isolation and characterization of novel toxins from snake venoms and identified a number of new toxins that belong to known family of toxins, but with different biological properties. We have also identified a few new families of snake venom toxins. The new toxins include (a ...
... We have been involved in isolation and characterization of novel toxins from snake venoms and identified a number of new toxins that belong to known family of toxins, but with different biological properties. We have also identified a few new families of snake venom toxins. The new toxins include (a ...

Eat more protein to keep weight off: study
... countries. The adults had already lost an average of 24 pounds after two months on a low-fat diet, and the researchers thought getting their family involved would help keep them on track. ...
... countries. The adults had already lost an average of 24 pounds after two months on a low-fat diet, and the researchers thought getting their family involved would help keep them on track. ...


English
... chains of proteins into smaller chains, which are in turn broken down into individual amino acids. These amino acids can then be rearranged into proteins that are found and used in our bodies. PowerPoint Slides 7 and 8. Have students create a flow chart on a sheet of paper showing how an enzyme brea ...
... chains of proteins into smaller chains, which are in turn broken down into individual amino acids. These amino acids can then be rearranged into proteins that are found and used in our bodies. PowerPoint Slides 7 and 8. Have students create a flow chart on a sheet of paper showing how an enzyme brea ...

Improved topology prediction using the terminal
... the most N-terminal TM-helix. In either case, the N-terminal helix is, on average, more hydrophobic than later helices (Hedin et al., 2010), see Figure 1. From a prediction point-of-view, this means that, at least one helix in a TM-protein, has to be more hydrophobic than almost any segment in a non ...
... the most N-terminal TM-helix. In either case, the N-terminal helix is, on average, more hydrophobic than later helices (Hedin et al., 2010), see Figure 1. From a prediction point-of-view, this means that, at least one helix in a TM-protein, has to be more hydrophobic than almost any segment in a non ...

Transient intracellular expression of chicken UCH-L3 and
... performed and peptide validator was used. Proteins were grouped, only PSMs with delta Cn better than: 0.1 were considered, and strict maximum parsimony principle was applied. Score versus Charge State filtering was also applied, in which the following minimal SEQUEST (XCorr) scores were considered f ...
... performed and peptide validator was used. Proteins were grouped, only PSMs with delta Cn better than: 0.1 were considered, and strict maximum parsimony principle was applied. Score versus Charge State filtering was also applied, in which the following minimal SEQUEST (XCorr) scores were considered f ...

1. introduction - International Journal of Computer Applications
... and this now become a standard in the field of bioinformatics. The Universal Protein Resource (Uniprot) is a central resource for protein sequences and functional information. The data set collected from UniProt KB (http://www.uniprot.org), for the functional information on proteins, are accurate, c ...
... and this now become a standard in the field of bioinformatics. The Universal Protein Resource (Uniprot) is a central resource for protein sequences and functional information. The data set collected from UniProt KB (http://www.uniprot.org), for the functional information on proteins, are accurate, c ...

Cambridge Isotope Laboratories, Inc. RefeRences 1. Shadforth, I.P.
... the mass spectrometer from liquid chromatography columns. In the mass spectrometer they are easily distinguished by their mass. Algorithms are then used to extract the light and heavy peptide ion chromatograms, which represent the peptide’s abundance. The light / heavy ratios are used to infer relat ...
... the mass spectrometer from liquid chromatography columns. In the mass spectrometer they are easily distinguished by their mass. Algorithms are then used to extract the light and heavy peptide ion chromatograms, which represent the peptide’s abundance. The light / heavy ratios are used to infer relat ...

Exercises in MBV-INF 4410/9410/9410A
... lowest isoform number. Also, if there are several entries for the same protein, select the one who has an accession starting with “NP_”, or alternatively with “XP_”. Retrieve the sequences in FASTA format, and paste them into the report. Shorten the titles of the sequences to contain only the protei ...
... lowest isoform number. Also, if there are several entries for the same protein, select the one who has an accession starting with “NP_”, or alternatively with “XP_”. Retrieve the sequences in FASTA format, and paste them into the report. Shorten the titles of the sequences to contain only the protei ...

The linear sequence of amino acids (primary structure) is able to coil
... The linear sequence of amino acids (primary structure) is able to coil and fold upon itself, resulting in 3D formations such as α-helices and β-sheets. These are held together by hydrogen bonding between amino acids. The term for these 3D formations is the secondary structure of the protein. ...
... The linear sequence of amino acids (primary structure) is able to coil and fold upon itself, resulting in 3D formations such as α-helices and β-sheets. These are held together by hydrogen bonding between amino acids. The term for these 3D formations is the secondary structure of the protein. ...
Protein mass spectrometry

Protein mass spectrometry refers to the application of mass spectrometry to the study of proteins. Mass spectrometry is an important emerging method for the characterization of proteins. The two primary methods for ionization of whole proteins are electrospray ionization (ESI) and matrix-assisted laser desorption/ionization (MALDI). In keeping with the performance and mass range of available mass spectrometers, two approaches are used for characterizing proteins. In the first, intact proteins are ionized by either of the two techniques described above, and then introduced to a mass analyzer. This approach is referred to as ""top-down"" strategy of protein analysis. In the second, proteins are enzymatically digested into smaller peptides using a protease such as trypsin. Subsequently these peptides are introduced into the mass spectrometer and identified by peptide mass fingerprinting or tandem mass spectrometry. Hence, this latter approach (also called ""bottom-up"" proteomics) uses identification at the peptide level to infer the existence of proteins.Whole protein mass analysis is primarily conducted using either time-of-flight (TOF) MS, or Fourier transform ion cyclotron resonance (FT-ICR). These two types of instrument are preferable here because of their wide mass range, and in the case of FT-ICR, its high mass accuracy. Mass analysis of proteolytic peptides is a much more popular method of protein characterization, as cheaper instrument designs can be used for characterization. Additionally, sample preparation is easier once whole proteins have been digested into smaller peptide fragments. The most widely used instrument for peptide mass analysis are the MALDI time-of-flight instruments as they permit the acquisition of peptide mass fingerprints (PMFs) at high pace (1 PMF can be analyzed in approx. 10 sec). Multiple stage quadrupole-time-of-flight and the quadrupole ion trap also find use in this application.