Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
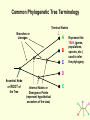
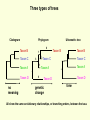
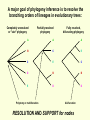
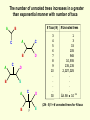
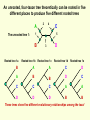
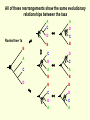
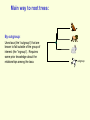
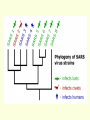
What is phylogenetic analysis and why should we perform it? Phylogenetic analysis has two major components: (1) Phylogeny inference or “tree building” the inference of the branching orders, and ultimately the evolutionary relationships, between “taxa” (entities such as genes, populations, species, etc.) (2) Analyzing change in traits (phenotypes, genes) using phylogenies as analytical frameworks for rigorous understanding of the evolution of various traits or conditions of interest Germline and somatic evolution included! Uses of Phylogenetics in the Study of Health & Disease (1) Evolutionary history of humans, between and within species (2) Analysis of evolution of phenotypic and genetic traits in humans, especially human-specific traits - evolved when, where, why, how (3) Evolution of parasites and pathogens, in relation to their hosts (us) (4) Evolution of cancer cell lineages, and somatic evolution more generally. (5) Study of adaptation in humans and other taxa What you will learn in this lecture (1) About phylogenies, terminology, what they are, how they work, ‘tree thinking’ (2) How to infer phylogenies (3) How we can use phylogenies to answer questions related to human adaptation, health and disease Common Phylogenetic Tree Terminology Terminal Nodes Branches or Lineages A B C D Ancestral Node or ROOT of the Tree Internal Nodes or Divergence Points (represent hypothetical ancestors of the taxa) E Represent the TAXA (genes, populations, species, etc.) used to infer the phylogeny Phylogenetic trees diagram the evolutionary relationships between the taxa Taxon B Taxon C Taxon A Taxon D No meaning to the spacing between the taxa, or to the order in which they appear from top to bottom. Taxon E This dimension either can have no scale (for ‘cladograms’), can be proportional to genetic distance or amount of change (for ‘phylograms’ or ‘additive trees’), or can be proportional to time (for ‘ultrametric trees’ or true evolutionary trees). ((A,(B,C)),(D,E)) = The above phylogeny as nested parentheses These say that B and C are more closely related to each other than either is to A, and that A, B, and C form a clade that is a sister group to the clade composed of D and E. If the tree has a time scale, then D and E are the most closely related. Three types of trees Cladogram Phylogram 6 Taxon B Taxon C Taxon A Taxon D no meaning 1 1 3 1 5 Ultrametric tree Taxon B Taxon B Taxon C Taxon C Taxon A Taxon A Taxon D Taxon D genetic change time All show the same evolutionary relationships, or branching orders, between the taxa. A major goal of phylogeny inference is to resolve the branching orders of lineages in evolutionary trees: Completely unresolved or "star" phylogeny Partially resolved phylogeny A A A B C E C E C D B B E D D Polytomy or multifurcation Fully resolved, bifurcating phylogeny A bifurcation RESOLUTION AND SUPPORT for nodes There are three possible unrooted trees for four taxa (A, B, C, D) Tree 1 Tree 2 Tree 3 A C A B A B B D C D D C Phylogenetic tree building (or inference) methods are aimed at discovering which of the possible unrooted trees is "correct". We would like this to be the “true” biological tree — that is, one that accurately represents the evolutionary history of the taxa. However, we must settle for discovering the computationally correct or optimal tree for the phylogenetic method of choice. The number of unrooted trees increases in a greater than exponential manner with number of taxa A # Taxa ( N) B C A B C A C B D D E A B C F D E 3 4 5 6 7 8 9 10 . . . . 30 # Unrooted trees 1 3 15 105 945 10,935 135,135 2,027,025 . . . . Å3.58 x 10 36 (2N - 5)!! = # unrooted trees for N taxa Inferring evolutionary relationships between the taxa requires rooting the tree: B To root a tree mentally, imagine that the tree is made of string. Grab the string at the root and tug on it until the ends of the string (the taxa) fall opposite the root: Root D Unrooted tree A A Note that in this rooted tree, taxon A is no more closely related to taxon B than it is to C or D. C B C D Rooted tree Root TIME Now, try it again with the root at another position: B C Root Unrooted tree D A A B C D Rooted tree TIME Root Note that in this rooted tree, taxon A is most closely related to taxon B, and together they are equally distantly related to taxa C and D. An unrooted, four-taxon tree theoretically can be rooted in five different places to produce five different rooted trees A The unrooted tree 1: 4 1 B Rooted tree 1a 2 Rooted tree 1b C 5 D 3 Rooted tree 1c Rooted tree 1d Rooted tree 1e B A A C D A B B D C C C C A A D D D B B These trees show five different evolutionary relationships among the taxa! All of these rearrangements show the same evolutionary relationships between the taxa Rooted tree 1a B A C D A C A D D C B B C D D C A A B B B B C D D A C A Main way to root trees: By outgroup: Uses taxa (the “outgroup”) that are known to fall outside of the group of interest (the “ingroup”). Requires some prior knowledge about the relationships among the taxa. outgroup Molecular phylogenetic tree building methods: Are mathematical and/or statistical methods for inferring the divergence order of taxa, as well as the lengths of the branches that connect them. There are many phylogenetic methods available today, each having strengths and weaknesses. Most can be classified as follows: COMPUTATIONAL METHOD Characters Distances DATA TYPE Optimality criterion Clustering algorithm PARSIMONY MAXIMUM LIKELIHOOD MINIMUM EVOLUTION UPGMA LEAST SQUARES NEIGHBOR-JOINING Types of data used in phylogenetic inference: Character-based methods: Use the aligned characters, such as DNA or protein sequences, directly during tree inference. Taxa Species Species Species Species Species ETC A B C D E Characters ATCGCTAGTCCTATAGTGCA ATCGCTAGTCCTATATTGCA TTCGCTAGACCTGTGGTCCA TTGACCAGACCTGTGGTCCG TTGACCAGTTCTGTGGTCCG ETC Similarity vs. Evolutionary Relationship: Similarity and relationship are not the same thing, even though evolutionary relationship is inferred from certain types of similarity. Similar: having likeness or resemblance (an observation) Related: genetically connected (an historical fact) Two taxa can be most similar without being most closely-related: 6 1 1 3 1 5 Taxon C Taxon A Taxon D Taxon B (eg HUMANS!) C is more similar in sequence to A (d = 3) than to B (d = 7), but C and B are most closely related (that is, C and B shared a common ancestor more recently than either did with A). Main computational approach: Optimality approaches: Use either character or distance data. First define an optimality criterion (minimum branch lengths, fewest number of events, highest likelihood), and then use a specific algorithm for finding trees with the best value for the objective function. Can identify many equally optimal trees, if such exist. Warning: Finding an optimal tree is not necessarily the same as finding the "true” tree. Random data will give you an ‘optimal’ (best ) tree! Parsimony methods: Optimality criterion: The ‘most-parsimonious’ tree is the one that requires the fewest number of evolutionary events (e.g., nucleotide substitutions, amino acid replacements) to explain the sequences. Advantages: • Are simple, intuitive, and logical (many possible by ‘pencil-and-paper’). • Can be used on molecular and non-molecular (e.g., morphological) data. • Can be used for character (can infer the exact substitutions) and rate analysis. • Can be used to infer the sequences of the extinct (hypothetical) ancestors. Disadvantages: • Not explicitly statistical • Can be fooled by high levels of parallel evolution Use parsimony to infer the optimal (best) tree Character-based methods: Use the aligned characters, such as DNA or protein sequences, directly during tree inference. Taxa Species Species Species Species Species A B C D E OUTGROUP ATCG ATCG TTCG TTGA TTGA Characters CTAGACCTATAGTGCA CTAGACCTATATTGCA CTAGACCTGTGGTCCA CCAGACCTGTGGTCCG CCAGTTGTGTGGTCCG TTAC CCATTTGTGTCCTCCG Infer maximum parsimony tree using first four characters Quality of trees (how likely it is that they reflect the one True Tree) can be evaluated in various ways (random data will give you a low-quality ‘best’ tree) We can Statistically Compare alternative trees, corresponding to specific biological hypotheses of the history of some set of lineages Time scales on trees: molecular clocks % genetic divergence Why such different profiles? Variation in mutation rate? 100% Fibrinopeptides 75% Hemoglobin 50% 25% Variation in selection. Genes coding for some molecules under very strong stabilizing selection. Cytochrome c Histone IV 300 600 900 1200 Time since divergence (Myr) 1500 Dates for calibrating molecular clocks can come from geology, fossils, or historical data From known ages of islands, for two genes Calibrating using fossil data chimps 6 substitutions humans whales 60 substitutions hippos 56 mya Calibrating from known dates of the ages of samples: for very fast-evolving taxa such as HIV Uses of Phylogenetics in the Study of Health & Disease (1) Evolutionary history of humans, between and within species (2) Analysis of evolution of phenotypic and genetic traits in humans, especially human-specific traits - evolved when, where, why, how (3) Taxonomy and evolution of parasites and pathogens, and evolution in relation to their hosts (4) Evolution of cancer cell lineages, and somatic evolution more generally. (5) Study of adaptation in humans and other taxa, via analysis of divergence and convergence EMERGING VIRUSES - THE GREATEST KNOWN HEALTH THREAT TO HUMANITY VIRUS - what IS it? Sequence it’s DNA and relate sequence to known viruses Evolution of SIV and HIV viruses: multiple transfers to humans, from chimps and from green monkeys SARS (severe acute respiratory syndrome) what causes it and where did it come from? HIV phylogeny within humans in different regions: Haiti as stepping stone to North America HIV evolves very rapidly WITHIN hosts, as a result of interactions with the immune system Can do phylogenetics: -Pathogens within individuals, -Pathogens between Individuals (eg in different or same regions) How originate? From other species? How spread? How does resistance to Antibiotics evolve in pathogens, & resistance to chemotherapeutic agents evolve in cancer? Cancer evolves genetically in the body during carcinogenesis, allowing the inference of ‘oncogenetic trees’ Cytogenetic data: Gains and losses of Chromosomal regions During evolution of cancers; Lose tumor suppressor gene copies, gain Oncogene copies Involves losses of heterozygosity and losses of imprinting Cancer Evolutionary Phylogenomics Compare primary cancer with metastatic tumors What you learned in this lecture (1) About phylogenies, terminology, what they are, how they work, ‘tree thinking’ (2) How to infer and evaluate phylogenies (3) How to use phylogenies to answer questions related to human adaptation, health and disease (viruses, cancer, etc) (4) How to THINK in terms of evolutionary trees (historical patterns of evolution), within and between species