Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Paracrine signalling wikipedia , lookup
Signal transduction wikipedia , lookup
Monoclonal antibody wikipedia , lookup
Ligand binding assay wikipedia , lookup
Point mutation wikipedia , lookup
Ribosomally synthesized and post-translationally modified peptides wikipedia , lookup
Green fluorescent protein wikipedia , lookup
Magnesium transporter wikipedia , lookup
Gene expression wikipedia , lookup
G protein–coupled receptor wikipedia , lookup
Ancestral sequence reconstruction wikipedia , lookup
Biochemistry wikipedia , lookup
Metabolomics wikipedia , lookup
Size-exclusion chromatography wikipedia , lookup
Expression vector wikipedia , lookup
Metalloprotein wikipedia , lookup
Interactome wikipedia , lookup
Homology modeling wikipedia , lookup
Fluorescence wikipedia , lookup
Bimolecular fluorescence complementation wikipedia , lookup
Protein–protein interaction wikipedia , lookup
Protein structure prediction wikipedia , lookup
Two-hybrid screening wikipedia , lookup
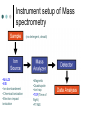
Methodology in protein science Yun-Ru (Ruby) Chen 陳韻如 Ph.D. The Genomics Research Center (office at 7th floor) [email protected] 2789-9930 ext 355 Protein synthesis in cell Incorporate non-natural amino acid in cell Protein Expression and Purification Volume 38, Issue 1, November 2004, Pages 37-44 Preparing protein samples What is the point? Endogenous proteins are not enough! Amplification Way to go Molecular cloning Heterogeneous expression Harvest and purification Hetero expression Bacteria: E. Coli Insect cells: baculo virus infection Mammalian cells: human suspension cells Bioorganism factory! Expression level, solubility, purification procedures, yield Detection/Quantification of proteins Quantification of pure protein UV absorbance: Tyr, Trp, Beer’s law A=ebc e=extinction coefficient Edhock equation. Abs280=1280*(# of Tyr)+2560*(# of Trp) • 1. 2. 3. 4. 5. 6. 7. 8. 9. • Specific Assays (functional based) Nonspecific assays 1. Catalytic activity Biuret ( rx peptide backbone) 2. Ligand binding Lowry( rx peptide backbone) 3. Antibody binding (western blot) Ninhydrin (rx free amino group) Fluorescamine (rx free amino group) Coomassie stain, noncovalent complex, ~10-7g detection Silver stain, <10-9g detection Direct blue Flamingo (fluorescence staining) SyproRuby (fluorescence staining) electrophoresis Agarose gel DNA SDS-PAGE sodium dodecyl sulfate-polyacrylamide gel electrophoresis (1 SDS molecule H-bonding with 2 residues) Native-PAGE chromatography Size-exclusion Ion-exchange Affinity hydrophobicity Size-exclusion chromatography Ion-Exchange Anion exchange Cation exchange Affinity Chromatography Common Fusion Tags and Purification Conditions Fusion Tag Immo bilize d Ligan d Glutath ione Stransfer ase(GS T) Binding Conditions Elution Conditions Available Formats Redu ced gluta thion e Neutral (physiologic) pH, and nondenaturing; glutathione must be reduced and GST must be active Free reduced glutathione at neutral pH (competitor) Prepacked column kits, spin cup column kits, SwellGel Discs, coated microplates Histidin etagged Chel ated Nick el or Coba lt Neutral (physiologic) pH without reducing or oxidizing agents; small tag must be accessible in fusion protein structure; high ionic strength and denaturants (chaotropes such as 8 M urea) compatible. >200 mM Imidazole, low pH, or strong chelators Prepacked column kits, spin cup column kits, SwellGel Discs, Swell- Gel Discs in 96well filter plates, coated microplates Maltos e Binding Protein (MBP) Dextr in Neutral (physiologic) pH and nondenaturing; NaCl added to reduce nonspecific binding Maltose at neutral pH (competitor) Gel slurry, coated microplates Green Fluores cent Protein (GFP) AntiGFP antib ody Neutral (physiologic) pH and nondenaturing Usual antibody/antigen elution buffers (e.g., low pH or chaotropic salts) Coated microplates Peptide synthesis The longer the more expensive Limitation at ~100 residues Relatively clean Methods to detect protein primary structure Protein sequencing By Edman degradation Phenylisothiocyanate (PITC) React with N- free amino acid Apply to chromatography Each amino acid eluted by Abs254nm (Error are cumulative) Instrument setup of Mass spectrometry (no detergent, desalt) •MALDI •ESI •Ion bombardment •Chemical ionization •Electron impact ionization •Magnetic •Quadrupole •Ion trap •TOF(Time of flight) •FT-MS Mass Spectrometry Combine with Edman degradation Combine with limited proteolysis (ex:Trypsin digestion) De novo sequencing is sometimes difficult (tandom mass) (LC/MS/MS) Advantage Protein doesn’t need high purity Picomoles of sample are required Also detect post-translational modification Methods to detect protein secondary structure Circular Dichroism Infrared spectrometry (FTIR) Light Waves farUV CD Circular dichroism (CD) is a form of spectroscopy based on the differential absorption of left- and right-handed circularly polarized light. It can be used to determine the structure of macromolecules (must be asymmetric) n -> pi* centered around 222 nm Part of pi -> pi* centered around 208 nm pi -> pi* centered around 190 nm n -> pi* involves non-bonding electrons of O of the carbonyl pi -> pi* involves the p-electrons of the carbonyl Far UV CD spectra Detect peptide backbone through pi bond formed by overlapping of 2 p orbital Alpha helix: min @222 and 208nm Beta sheet: min @216nm, max @195nm Random coil: decreasing signal below 200nm, slight increase @218nm Signal to noise ratio: Protein concentration, Path length, salt in buffer, response time Units: ellipticity (θ), 32.98 θ = 33.98 ΔAbs Ellipticity: millidegree Molar ellipticity ([θ]) is CD corrected for concentration. molar elliplicity are historical (deg cm2/dmol) the sample concentration (g/L), cell pathlength (cm), and the molecular weight (g/mol) must be known % alpha-helix = (-[θ]222nm +3000)/39000 Biochemistry. 39, 11657-11666, 2000 Secondary Structure Prediction needs spectra down to at least 200nm (some need 178nm) Infrared spectra The frequencies with which bonded atoms vibrate relative to each other determine the vibrational spectrum of a molecule. High background of water. Often use D2O Amide I is the most sensitive Spectra need to be deconvoluted Methods to detect protein tertiary/quaternary structural changes Size exclusion chromatography Fluorescence spectroscopy Near UV Circular dichroism Analytical Ultracentrifugation (AUC) NMR X-ray crystallography Fluorescence Fluorescence Wavelength scan Fluorescence Anisotropy Fluorescence correlation spectrum Fluorescence Life Time Fluorescence energy transfer Highly sensitive Small amount (ug) Give total conformational information Fluorescence quantum yield=(# of (3))/(# of (1)) Jablonski diagram fluorescence intensity (cps/uA) 25E+06 20E+06 15E+06 10E+06 50E+05 0 300 cps/uA / Wavelength (nm) 350 400 450 500 550 600 emission wavelength, nm File # 2 = Y-M2A λem>λex 650 700 750 Stern-Volmer equation, Ksv=t0kq Fluorescence instrumentation Fluorescence Anisotropy Polarization techniques can provide average size and shape of rotating fluorophores and macromolecules Polarization (P) = (Iv - Ih) / (Iv+ Ih) Anisotropy (r) = (Iv - Ih) / (Iv+ 2Ih) where Iv is the intensity parallel to the excitation plane and Ih is the emission perpendicular to the excitation plane. They are interchangeable quantities and only differ in their normalization. Polarization P ranges from –0.33 to +0.5 while the range for anisotropy r is –0.25 to +0.4. Fluorescence energy transfer A donor chromophore in its excited state can transfer energy by a nonradiative, long-range dipole-dipole coupling mechanism to an acceptor chromophore in close proximity (typically <10nm). Fluorescence correlation spectrum Using confocal or two photon microscopy, light is focused on a sample and the measured fluorescence intensity fluctuations (due to diffusion, chemical reactions, aggregation, etc.). FCS is the fluorescent counterpart to dynamic light scattering, FCS obtains quantitative information such as diffusion coefficients, hydrodynamic radii, average concentrations, kinetic chemical reaction rates. Fluorescence Life Time The fluorescence lifetime refers to the average time the molecule stays in its excited state before emitting a photon. Fluorescence typically follows first-order kinetics: where [S]t is the concentration of excited state molecules at time t, [S]0 is the initial concentration and Γ is the decay rate or the inverse of the fluorescence lifetime. This is an instance of exponential decay. [S]t = [S]0 exp (Γ* t) What Information Can AUC Provide? Sedimentation equilibrium Molecular weight Stoichiometry Oligomerization (Kd from 10-3-10-8 M) The Sedimentation velocity Shape and size Number of species Diffusion constant first model was built in 1924 by Theodore (The) Svedberg. Diagram of a Sedimentation Experiment Fc: centrifugal force Fb: buoyancy Fd: frictional force Sedimentation Equilibrium Change in Concentration Over Time Basic Theory of Sedimentation Equilibrium C C: concentration rm r: radius w: angular velocity JS: Flux of sedimentation JD: Flux of diffusion (Fick’s Law) s: sedimentation coefficient rb c(r ) = c0e r 2 r02 ( = ) 2 2 Curve fitting with assumed model or plotting method Different Cases for Sedimentation Equilibrium 1. 2. 3. 4. 5. 6. Single species (monomer) self-association (homooligomer) Heterogeneous mixtures (heterooligomer) Multiple component versus multiple species Heterogeneous associations Non ideally Plotting the residual as a function of radial distance Sedimentation Velocity Determining the rate of movement of a solute under a centrifugal field The centrifugal force on the particle (solute) is equal to the friction of the particle. dr M b w r = M (1 )w r = fv = f dt 2 2 Comparison to Other Useful Techniques AUC Mass Spectrometry MicroCalorimetry Fluorescence spectrometry, CD, Light scattering Solution MW MW Thermodynamics (DG, DH, DC) Enzyme kinetics, 2nd structures, solution mass Stoichiometry, Assembly Model Non covalent interactions Molar ratio Stoichiometry Thermodynamics( D G) Stoichiometry Folding stability Folding stability Conformational changes, shape Epitope mapping Conformational changes Conformational changes Kd 10-3~10-8 Identify unknowns Kd 10-3~10-11; Kd 10-6~10-11 M 10-6~10-20 M Methods to detect atomic level protein structure Amount X-ray Crystallography NMR 10mg/ml 10mg/ml pro Atomic level Large protein (50-100kD) con Magic required Rigid structure Mostly native state Atomic level Size limit Flexible proteins (<50kD) Dynamics Over large time scale EM Data derived from physical techniques for probing structure, the interpretation is not unambiguous and entails assumptions and approximations often depending upon knowledge of the proteins from other sources (biology) X-ray Crystallography Bragg’s Law The interference is constructive when the phase shift is a multiple to 2π; this condition can be expressed by Bragg's law: Crystallizing proteins Both entail a droplet containing purified protein, buffer, and precipitant being allowed to equilibrate with a larger reservoir containing similar buffers and precipitants in higher concentrations. Initially, the droplet of protein solution contains an insufficient concentration of precipitant for crystallization, but as water vaporizes from the drop and transfers to the reservoir, the precipitant concentration increases to a level optimal for crystallization. NMR spectroscopy (nuclear magnetic resonance) Determine internuclear distances by measuring perturbations between assigned resonances from atoms in the protein in solution 1D NMR Proton: 1H, Isotope labeled carbon 13C and nitrogen 15N, 19F Different nuclei in a protein absorb electromagnetic energy (resonance) at different frequencies because their local electromagnetic environment differ Useful parameters Chemical shift (freq) Chemical structure Spin-spin coupling (through bond) 3J Signal intensity Concentration NHa torsion angle for structural constraint Nuclear overhauser effect (NOE) Intermolecular distance (through space) Relaxation time (T1, T2) Motional dynamics Signal linewidth dynamics 2D NMR Electron Microscope Electron beam is stronger than X-ray. No need for 3D crystal Achieve atomic level resolution electrons interact more strongly with atoms than X-rays Phase problem less severe But could destroy sample a) b) c) d) Wire Ribbon Ball and Stick space filling as a sphere of van der Waals radius e) surface representation GRASP image topology of protein surface (red negative, blue positive) Protein structure determination methods High resolution X-ray crystallography | NMR | Electron crystallography Medium Cryo-electron microscopy | Fiber diffraction | Mass spectrometry resolution | SAXS Spectroscopic NMR | Circular dichroism | Absorbance | Fluorescence | Fluorescence anisotropy Translational Analytical ultracentrifugation | Size exclusion chromatography | Diffusion Light scattering | NMR Rotational Fluorescence anisotropy | Flow birefringence | Dielectric Diffusion relaxation | NMR Chemical Hydrogen-deuterium exchange | Site-directed mutagenesis | Chemical modification Thermodynamic Equilibrium unfolding Computational Protein structure prediction | Molecular docking