Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Molecular cloning wikipedia , lookup
Zinc finger nuclease wikipedia , lookup
Molecular ecology wikipedia , lookup
Bisulfite sequencing wikipedia , lookup
Proteolysis wikipedia , lookup
Transposable element wikipedia , lookup
Metalloprotein wikipedia , lookup
Catalytic triad wikipedia , lookup
Genetic code wikipedia , lookup
Vectors in gene therapy wikipedia , lookup
Promoter (genetics) wikipedia , lookup
Genomic library wikipedia , lookup
Gene expression wikipedia , lookup
Endogenous retrovirus wikipedia , lookup
Silencer (genetics) wikipedia , lookup
Community fingerprinting wikipedia , lookup
Non-coding DNA wikipedia , lookup
Two-hybrid screening wikipedia , lookup
Deoxyribozyme wikipedia , lookup
Point mutation wikipedia , lookup
Protein structure prediction wikipedia , lookup
Ancestral sequence reconstruction wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
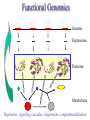
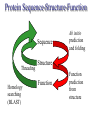
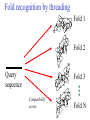
C E N T R E F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U 1-Month Practical Master Course Genome Analysis Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU) Vrije Universiteit Amsterdam The Netherlands www.ibivu.cs.vu.nl [email protected] Chemistry Biology Molecular biology Mathematics Statistics Bioinformatics Computer Science Informatics Medicine Physics Biological Sequence Analysis Pair-wise sequence alignment Residue exchange matrices Multiple sequence alignment Phylogeny C E N T R E F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U DNA sequence .....acctc tcccagatgg ggcccaggac ttggtgacac caaatcttgt gagcccaaat gcccagagcc ccggtgccca ttcctcttcc cccggacccc ccacgaagac ggcgtggagg agcagtacaa cgtcctgcac tgcaaggtct cctggtcaaa tgggagagca cgcctcccat cagcaagctc aacatcttct accgctacac ctgtgcaaga gtcctgtccc tggggaagcc aactcacaca gacacacctc cttgtgacac caaatcttgt gcacctgaac ccccaaaacc tgaggtcacg ccnnnngtcc tgcataatgc cagcacgttc caggactggc ccaacaaagc ggcttctacc atgggcagcc gctggactcc accgtggaca catgctccgt gcagaagagc acatgaaaca aggtgcacct tccagagctc tgcccacggt ccccgtgccc acctccccca gacacacctc tcttgggagg caaggatacc tgcgtggtgg agttcaagtg caagacaaag cgtgtggtca tgaacggcaa aaccaagtca ccagcgacat ggagaacaac gacggctcct agagcaggtg gatgcatgag ctctc..... nctgtggttc gcaggagtcg aaaaccccac gcccagagcc acggtgccca tgcccacggt ccccgtgccc accgtcagtc cttatgattt tggacgtgag gtacgtggac ctgcgggagg gcgtcctcac ggagtacaag gcctgacctg cgccgtggag tacaacacca tcttcctcta gcagcagggg gctctgcaca Genome size Organism Number of base pairs X-174 virus 5,386 Epstein Bar Virus 172,282 Mycoplasma genitalium 580,000 Hemophilus Influenza 1.8 106 Yeast (S. Cerevisiae) 12.1 106 Human 3.2 109 Wheat 16 109 Lilium longiflorum 90 109 Salamander 100 109 Amoeba dubia 670 109 Three main principles • DNA makes RNA makes Protein • Structure more conserved than sequence • Sequence Structure Function Functional Genomics Genome Expressome Proteome TERTIARY STRUCTURE (fold) TERTIARY STRUCTURE (fold) Metabolome Regulation, signalling cascades, chaperonins, compartmentalisation How to go from DNA to protein sequence A piece of double stranded DNA: 5’ attcgttggcaaatcgcccctatccggc 3’ 3’ taagcaaccgtttagcggggataggccg 5’ DNA direction is from 5’ to 3’ How to go from DNA to protein sequence 6-frame translation using the codon table (last lecture): 5’ attcgttggcaaatcgcccctatccggc 3’ 3’ taagcaaccgtttagcggggataggccg 5’ Evolution and three-dimensional protein structure information Isocitrate dehydrogenase: The distance from the active site (in yellow) determines the rate of evolution (red = fast evolution, blue = slow evolution) Dean, A. M. and G. B. Golding: Pacific Symposium on Bioinformatics 2000 Protein Sequence-Structure-Function Sequence Threading Homology searching (BLAST) Ab initio prediction and folding Structure Function Function prediction from structure Widely used tool for homology detection: PSI-BLAST • Heuristic tool to cut down computations required for database searching (~1M sequences in DB) • Sensitivity gained by iteratively finding hits (local alignments) and repeating search Q hits T PSSM DB Threading Template sequence Compatibility score + Query sequence Template structure Threading Template sequence Compatibility score + Query sequence Template structure Fold recognition by threading Fold 1 Fold 2 Query sequence Fold 3 Compatibility scores Fold N Bioinformatics “Nothing in Biology makes sense except in the light of evolution” (Theodosius Dobzhansky (1900-1975)) “Nothing in bioinformatics makes sense except in the light of Biology” Divergent evolution Ancestral sequence: ABCD ACCD (B C) ACCD AB─D or ABD (C ø) mutation deletion ACCD A─BD Pairwise Alignment Divergent evolution Ancestral sequence: ABCD ACCD (B C) ACCD AB─D true alignment or ABD (C ø) mutation deletion ACCD A─BD Pairwise Alignment Mutations under divergent evolution (a) G (b) G Ancestral sequence G Sequence 1 A One substitution one visible Sequence 2 1: ACCTGTAATC 2: ACGTGCGATC * ** D = 3/10 (fraction different sites (nucleotides)) C (c) G C Two substitutions one visible (d) G G A A Two substitutions none visible A Back mutation not visible G Convergent evolution • Often with shorter motifs (e.g. active sites) • Motif (function) has evolved more than once independently, e.g. starting with two very different sequences adopting different folds • Sequences and associated structures remain different, but (functional) motif can become identical • Classical example: serine proteinase and chymotrypsin Serine proteinase (subtilisin) and chymotrypsin • Different evolutionary origins, no sequence similarity • Similarities in the reaction mechanisms. Chymotrypsin, subtilisin and carboxypeptidase C have a catalytic triad of serine, aspartate and histidine in common: serine acts as a nucleophile, aspartate as an electrophile, and histidine as a base. • The geometric orientations of the catalytic residues are similar between families, despite different protein folds. • The linear arrangements of the catalytic residues reflect different family relationships. For example the catalytic triad in the chymotrypsin clan (SA) is ordered HDS, but is ordered DHS in the subtilisin clan (SB) and SDH in the carboxypeptidase clan (SC). A protein sequence alignment MSTGAVLIY--TSILIKECHAMPAGNE-------GGILLFHRTHELIKESHAMANDEGGSNNS * * * **** *** A DNA sequence alignment attcgttggcaaatcgcccctatccggccttaa att---tggcggatcg-cctctacgggcc---*** **** **** ** ****** What can sequence tell us about structure (HSSP) Sander & Schneider, 1991 Searching for similarities What is the function of the new gene? The “lazy” investigation (i.e., no biologial experiments, just bioinformatics techniques): – Find a set of similar protein sequences to the unknown sequence – Identify similarities and differences – For long proteins: identify domains first Evolutionary and functional relationships Reconstruct evolutionary relation: •Based on sequence -Identity (simplest method) -Similarity •Homology (common ancestry: the ultimate goal) •Other (e.g., 3D structure) Functional relation: Sequence Structure Function Searching for similarities Common ancestry is more interesting: Makes it more likely that genes share the same function Homology: sharing a common ancestor – a binary property (yes/no) – it’s a nice tool: When (an unknown) gene X is homologous to (a known) gene G it means that we gain a lot of information on X: what we know about G can be transferred to X as a good suggestion. Biological definitions for related sequences Homologues are similar sequences in two different organisms that have been derived from a common ancestor sequence. Homologues can be described as either orthologues or paralogues. Orthologues are similar sequences in two different organisms that have arisen due to a speciation event. Orthologs typically retain identical or similar functionality throughout evolution. Paralogues are similar sequences within a single organism that have arisen due to a gene duplication event. Xenologues are similar sequences that do not share the same evolutionary origin, but rather have arisen out of horizontal transfer events through symbiosis, viruses, etc. How to evolve Important distinction: • Orthologues: homologous proteins in different species (all deriving from same ancestor) • Paralogues: homologous proteins in same species (internal gene duplication) • In practice: to recognise orthology, bi-directional best hit is used in conjunction with database search program (this is called an operational definition) So this means … Source: http://www.ncbi.nlm.nih.gov/Education/BLASTinfo/Orthology.html Example today: Pairwise sequence alignment needs sense of evolution Global dynamic programming MDAGSTVILCFVG M D A A S T I L C G S Evolution Search matrix MDAGSTVILCFVGMDAAST-ILC--GS Amino Acid Exchange Matrix Gap penalties (open,extension) How to determine similarity Frequent evolutionary events at the DNA level: 1. Substitution 2. Insertion, deletion 3. Duplication 4. Inversion We will restrict ourselves to these events nucleotide oneletter code A DNA sequence alignment attcgttggcaaatcgcccctatccggccttaa att---tggcggatcg-cctctacgggcc---*** **** **** ** ****** A protein sequence alignment MSTGAVLIY--TSILIKECHAMPAGNE-------GGILLFHRTHELIKESHAMANDEGGSNNS * * * **** *** amino acid oneletter code Dynamic programming Scoring alignments – Substitution (or match/mismatch) • DNA • proteins – Gap penalty • Linear: gp(k)=ak • Affine: gp(k)=b+ak • Concave, e.g.: gp(k)=log(k) The score for an alignment is the sum of the scores over all alignment columns Dynamic programming Scoring alignments Sa,b = s(a , b ) - l i j gp(k) = gapinit + kgapextension k Nk gp(k ) affine gap penalties DNA: define a score for match/mismatch of letters Simple: A C G T A 1 -1 -1 -1 C -1 1 -1 -1 G -1 -1 1 -1 T -1 -1 -1 1 Used in genome alignments: A C G T A 91 -114 -31 -123 C -114 100 -125 -31 G -31 -125 100 -114 T -123 -31 -114 91 Dynamic programming Scoring alignments T D W V T A L K T D W L - - I K 2020 10 Amino Acid Exchange Matrix 1 Affine gap penalties (open, extension) Score: s(T,T)+s(D,D)+s(W,W)+s(V,L)-Po-2Px + +s(L,I)+s(K,K)