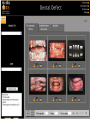
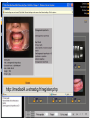
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
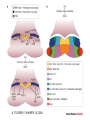
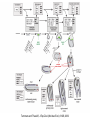
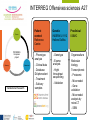
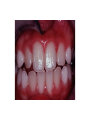
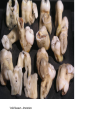
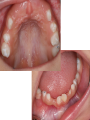
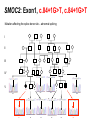
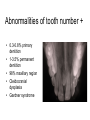
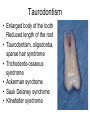
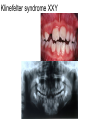
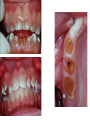
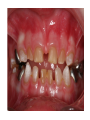
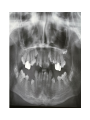
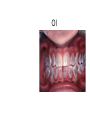
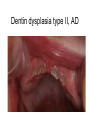
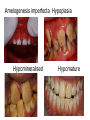
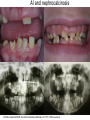
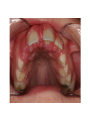
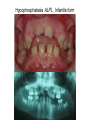
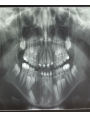
Teeth and syndromes A. BLOCH-ZUPAN Faculté de Chirurgie Dentaire, Université de Strasbourg; Centre de Référence pour les manifestations bucco-dentaires des maladies rares; Pôle de Médecine et Chirurgie Bucco-Dentaires, Hôpitaux Universitaires Strasbourg, France; IGBMC (Institut de Génétique et de Biologie Moléculaire et Cellulaire), INSERM U964, UMR7104 CNRS, Illkirch; ICS, Illkirch, France Orodental anomalies are one aspect of rare diseases or syndromes Prevalence < 1/2000 80% genetic Rare diseases 4 millions people in France 7000 syndromes 8000 25 millions people in Europe +900 with orodentofacial anomalies 750 with cleft lip and/or palate è Few information è Adequate care and management, diagnostic issues è The oral cavity a door towards diagnosis treatment and patient management 65 % rare diseases are severe Williams syndrome - 1/10 000 - Cardiac Malformation - Facial Dysmorphism Osteogenesis Imperfecta - 1/100 000 - DI Lesch-Nyhan Disease - 1/1 000 000 - Enzymatic deficit (purine) -Motor deficit - Self mutilation Specific orodental manifestations Non specific caries, periodontal diseases, specific management The oral cavity : a door towards development and pathology Odontogenesis • • • • • • • • • • • Origin of dental cells Determination of tooth region Determination of tooth identity Determination of tooth shape Cytodifferentiation of Od and Am Dentin and Enamel Root development, Eruption Alveolar bone formation Stem cells Palatogenesis EMT Bone formation and metabolism Anomalies • • • Number Shape Size • • Structure Colour • • Root Eruption Cleft L/P Embryonic origin of dental cells • Enamel organ oral ectoderm, ameloblasts • Dental mesenchyme (dental papilla, odontoblasts, periodontium, bone of attachement cells) derives from cranial neural crest – Prosencephalon + rostral mesencephalon populating the periocular and frontonasal regions contributes to the upper incisors – Rostral rhombencephalon (Rhombomeres 1 and 2) and caudal mesencephalon populates the first maxillo-mandibular pharyngeal arch thus contributing to the lower incisors, the lower and upper molars. MES RHO PRO * * A. TUCKER, P. SHARPE, (5) 2004 Tummers and Thesleff, J Exp Zool (Mol dev Evol) 312B, 2009 Molar Incisor Molar Premolar Canine Incisor Fleischmannova J. et al., Eur J Oral Sci. Feb;116(1):1-10. (2008) Osteopetrosis / Ae2 - Increased bone density - Diffuse and focal sclerosis of varying severity - Modelling defects at metaphyses - Pathological fractures - Osteomyelitis - Dental abnormalities: tooth eruption defects INTERREG Offensives sciences A27 France INTERREG European project Germany Switzerland Translational Research Patient centred Reference Centre Genetic INSERM U 1112 Helène Dollfus - Phenotype analysis - Clinical data - Database D4/phenodent - Treatment - Salivary samples - Genotype - Exome analysis - Hight throughput sequencing - Validation Preclinical IGBMC Organoculture Molecular biology Transcriptomic - Proteomic - Mice model - Gene validation - Mice model analysis by microCT - SEM Région Alsace, Ministerium für Wissenschaft, Forschung und Kunst Baden Württemberg, Ministerium für Bildung, Wissenschaft, Weiterbildung und Kultur des Landes RheinlandPfalz, Université de Strasbourg, Hôpitaux Universitaires de Strasbourg, CERBM – IGBMC, EA3949 - Laboratoire de Génétique Médicale, Universitätsklinikum Freiburg, Universität Heidelberg, Hypophosphatasie Europe, O b e r r h e i n i s c h e Zahnärtztegesellschaft Genes transcribed at mouse E14.5 cap stage - Transcriptome atlas 19872 genes expressed at cap stage E14.5 387 genes involved in a disease with « teeth abnormalities » OMIM, Manteia 405 different diseases Among 19872 genes expressed at mouse tooth cap stage, 387 were encountered in 405 diseases with «teeth abnormalities » Molars and incisors: show your microarray IDs. Laugel-Haushalter V, Paschaki M, Thibault-Carpentier C, Dembelé D, Dollé P, Bloch-Zupan A. BMC Res Notes. 2013 Mar 26;6:113. Vrolik Museum. Amsterdam. Abnormalities of tooth number • Fewer teeth – Hypodontia (≤ 6 missing teeth (permanent) – Oligodontia (>6 congenitally missing teeth) – Anodontia (no teeth) • More teeth – Supernumerary – Hyperdontia Frequencies • Hypodontia • Primary dentition • Permanent dentition – Maxillary lateral incisor – Maxillary second premolar – Mandibular second premolar – Third molar – Maxillary central incisor – Ca, 1st PreMo, 1st Mo, 2nd Mo 0.1-0.7% 2.3-9.6% 27% 15% 32% 25% 0.05% 1% • Missing of a primary tooth is followed by the absence of the permanent successor in 75-80% of cases Mucchielli ML, Mitsiadis TA, Raffo S, Brunet JF, Proust JP, Goridis C. Dev Biol. 1997 Sep 15;189(2):275-‐84. Missing teeth and MSX1 mutations AD, 4p16.1 2 1 3 3 1 2nd premolar > 3rd molar > 1st premolar > 2 Van den Boogaard et al., 2000 § CL/P The same MSX1 mutation can cause teeth agenesis and CL/P Jumlongras et al., 2001 § Witkop syndrome : AD missing teeth and nail defects Stockton et al., 2000 V.5 (13 ans) Missing teeth and PAX9 mutations 1 2 3 3rd molar > 2nd molar > 1st molar> 3 2 1 Rieger syndrome II:2 R PITX2, AD, 4q25-q26 III:2 FOXC1, AD, 6p25 IV:1 IV:1 A III:2 D B E C F A novel homeobox mutation in the PITX2 gene in a family with Axenfeld-Rieger syndrome associated with brain, ocular, and dental phenotypes. Idrees F, Bloch-Zupan A, Free SL, Vaideanu D, Thompson PJ, Ashley P, Brice G, Rutland P, Bitner-Glindzicz M, Khaw PT, Fraser S, Sisodiya SM, Sowden JC. Am J Med Genet B Neuropsychiatr Genet. 2006 Mar 5;141B(2):184-91. Pathway http://david.abcc.ncifcrf.gov/kegg.jsp?path=hsa04310$Wnt signaling p... Pathway:Wnt signaling pathway Pathway information generated by KEGG. List genes are shown in red DAVID Gene Name C-terminal binding protein 1 C-terminal binding protein 2 Stop Blinking Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Lammi L, Arte S, Somer M, Jarvinen H, Lahermo P, Thesleff I, Pirinen S, Nieminen P. Am J Hum Genet. 2004 May;74(5):1043-50. WNT10A missense mutation associated with a complete odonto-onycho-dermal dysplasia syndrome (AR, 2q35 ). Nawaz S, Klar J, Wajid M, Aslam M, Tariq M, Schuster J, Baig SM, Dahl N. Eur J Hum Genet. 2009 Dec;17(12):1600-5. Epub 2009 May 27. WNT10A non syndromic hypodontia 16% of patients with 1-3 missing teeth and 51.6 % (16/31) of patients with 4 or more missing teeth Song et al, Hum Genet. 2013 : The Ectodysplasin and NFkappaB signalling pathways in odontogenesis. Courtney JM, Blackburn J, Sharpe PT. Arch Oral Biol. 2005 Feb;50(2):159-63. Review. X linked Hypohydrotic ectodermal dysplasia with EDA gene mutation (exons 4-9 deletion). F Clauss CR Hypohydrotic ectodermal dysplasia with EDAR gene mutation F Clauss CR Incontinentia Pigmenti NEMO, IKKgamma, Xq28 EEC (ED, Ectrodactyly, CP) P63, 3q27 Missing: 83,12,13,14,14,22,23,24,25,41,42,4344 45,47,31,32,33,34,35,37 EEC M Holder-Espinasse Lille, France Nicolas Chassaing, Toulouse, France Mutations in different components of FGF signaling in LADD syndrome. Rohmann E, Brunner HG, Kayserili H, Uyguner O, Nürnberg G, Lew ED, Dobbie A, Eswarakumar VP, Uzumcu A, UlubilEmeroglu M, Leroy JG, Li Y, Becker C, Lehnerdt K, Cremers CW, Yüksel-Apak M, Nürnberg P, Kubisch C, Schlessinger J, van Bokhoven H, Wollnik B. Nat Genet. 2006 Apr;38(4):414-7 Kallman syndrome, FGFR1, 8p11.2-p11.1 Central hypogonadism Lack of sense of smell Renal aplasia Deafness Syndactyly Cleft lip/palate Tooth agenesis I Bailleul-Forestier Toulouse Isolated tooth agenesis, PreMo Viera, 2007 Modulation of Fgf3 dosage in mouse and men mirrors evolution of mammalian dentition Cyril Charles, Vincent Lazzari, Paul Tafforeau, Thomas Schimmang, Mustafa Tekin, Ophir Klein and Laurent Viriot PNAS, 106, 52, 2009 Fgf3+/+ Fgf3+/– Fgf3–/– Oral-facial-digital syndrome Type I OFD1(Cxorf5/71-7a), Xp22 M-C Manière CR Oligodontia in Johanson-Blizzard syndrome, UBR1, AR, 15q15.2 • Characteristic facies, severe mental and somatic retardation, sensorineural hearing loss, malabsorption due to pancreatic insuficiency • Microdontia • All permanent teeth absent except for the first permanent molars, incisors Bloom syndrome • #210900 - 15q26.1 - RECQL3 • DNA helicase -ATP-dependent RNA or DNA unwinding Cockayne syndrome (Type A – CSA; or CS Type I OMIM #216400) (Type B – CSB; or CS Type II OMIM #133540) ERCC6, ERCC8 A possible cranio-oro-facial phenotype in Cockayne syndrome. Bloch-Zupan A, Rousseaux M, Laugel V, Schmittbuhl M, Mathis R, Desforges E, Koob M, Zaloszyc A, Dollfus H, Laugel V. Orphanet J Rare Dis. 2013 Jan 14;8(1):9. Unique phenotype 6q27 3Mb Affected Rela+ves Non affected Haplotypes / A single shared homozygous region Bioinformatics Phenotype Bibliography Hypothèses 69 Genes Transcriptome Atlas Homozygous area Genes Functions Phylogeny Proteins Expression pattern Interactions Candidates 2 Genes SMOC2, SPARC related modular calcium binding 2 – 226kb - 13 Exons DACT2, dapper, antagonist of beta-catenin, homolog 2 –26.93 kb - 4 Exons SMOC2: Exon1, c.84+1G>T, c.84+1G>T Mutation affecting the splice donor site – abnormal splicing I II III IV IV:3 IV:9 IV:4 V V:1 V:2 V:4 V:3 IV:4 IV:3 V:1 V:2 V:3 IV:10 V:5 V:6 IV:9 V:4 IV:10 V:5 V:6 SMOC2 SPARC related modular calcium binding 2 This gene is coding for - a member of the SPARC (secreted protein acidic and rich in cysteine/ osteonectin/BM-40) family - expressed during embryonic development - an extracellular matrix protein binding calcium - facilitating matrix formation, proliferation, cell migration (endothelial) and angiogenesis SMOC proteins family The domain structure of SMOC1 and SMOC2 is composed of 5 domains, one KAZAL domain (a serine protease inhibitor), two thyroglobulin type I repeats (predicted to be inhibitors of cysteine proteases and binding partners of heparin) and a calcium-binding domain, which is conserved throughout the evolution. SMART, eggNOG, Orthoinspector, AQUA, GBLOCK, PhyML, ReadSEQ, iTOL 14q24.2 A C B P P P T Mo T Mo MC S D S E Mo1 Mo2 Mo1 F G ★ H ★ Inc Inc Tooth development Zebrafish versus Mouse [Borday-Birraux et al (2006) Evol Dev 8: 130-141] Christelle ETARD et Uwe STRAHLE, Karlsruher Institut für Technologie (KIT), Institut für Toxikologie und Genetik (ITG), Allemagne SMOC2 exome sequencing IntegraGen exome : 69 genes analyzed using VaRank (V. Geoffroy) - 81 substitutions (47 intronic, or in the untranslated regions; 22 synonymous; and 12 missense, of which all were SNPs) - 7 deletions (all intronic and four SNPs) - 1 insertion (all intronic and one SNP). Good coverage of other SMOC2 exons SMOC2 Exome CTRL Affected No coverage of the mutation region c.84+1G>T Homozygosity mapping and candidate prioritization identify mutations, missed by whole-exome sequencing, in SMOC2, causing major dental developmental defects. Bloch-Zupan A, Jamet X, Etard C, Laugel V, Muller J, Geoffroy V, Strauss JP, Pelletier V, Marion V, Poch O, Strahle U, Stoetzel C, Dollfus H. Am J Hum Genet. 2011 Dec 9;89(6):773-81. Recessive oligodontia linked to a homozygous loss-of-function mutation in the SMOC2 gene. Alfawaz S, Fong F, Plagnol V, Wong FS, Fearne J, Kelsell DP.Arch Oral Biol. 2013 a homozygous mutation, NM_022138.2: c.681T>A, in the SMOC2 gene changing a cysteine to a premature stop termination codon at codon 227 (p.C227X) in exon 8 Abnormalities of tooth number + • 0.3-0.8% primary dentition • 1-3.5% permanent dentition • 98% maxillary region • Cleidocranial dysplasia • Gardner syndrome Cleidocranial dysplasia, Runx2, AD, 6p21 Gardner syndrome Familial adenomatous polyposis , APC, negative regulator of Wnt, AD, 5q21-q22 several well-defined opacities around the periphery of the mandible Gardner's syndrome - a case report. Payne M, Anderson JA, Cook J. Br Dent J. 2002 Oct 12;193(7):383-4. Nance-Horan syndrome, NHS, Xp22.13 • X-linked cataract with dental anomalies • Number – Supernumerary – Agenesis • Shape – Inc screwdriver-shaped, tapering – Cingular cusps – Premolars, molars rounded – Taurodontism, wide pulp chambers • Eruption, Impacted teeth Abnormalities of tooth shape • Double formation • 0.14-3% • 0.2% • KBG Abnormalities of tooth shape • Talon cusp (T cingulum) • OFDII, Rubinstein Taybi syndromes Tooth shape/size anomalies • Dens invaginatus, dens in dente • 0.25-10%, 12 or 22 6-10%, bilateral 43% • Dens invaginatus and deafness • Taurodontism, microdontia and dens invaginatus • Postaxial polydactyly-dental vertebral syndrome Taurodontism • Enlarged body of the tooth Reduced length of the root • Taurodontism, oligodontia, sparse hair syndrome • Trichodento-osseous syndrome • Ackerman syndrome • Sauk Delaney syndrome • Klinefelter syndrome Klinefelter syndrome XXY Case report: Macrodont mandibular second premolars, a hereditary dental anomaly. Kyriazidou A, Haider D, Mason C, Parekh S, Bloch-Zupan A. Eur Arch Paediatr Dent. 2013 Jun 5. Ekman-Westborg and Julin syndrome Multiple macrodontic multituberculism. Benjamin MR, Rodrigo FS, Gorlin RJ. Am J Med Genet A. 2003 Jul 15;120A(2):283-5. Otodental syndrome • • • • • Sensorineural hearing loss Large globe shaped molars (globodontia) Supernumerary microdont teeth Taurodontism Delayed development and eruption Oto-dental pedigrees UK Belgium Brazil SNP genome scanning localizes oto-dental syndrome to chromosome 11q13 and microdeletions at this locus implicate FGF3 in dental and inner-ear disease and FADD in ocular coloboma. Gregory-Evans CY, Moosajee M, Hodges MD, Mackay DS, Game L, Vargesson N, Bloch-Zupan A, Rüschendorf F, Santos-Pinto L, Wackens G, Gregory-Evans K. Hum Mol Genet. 2007 Oct 15;16(20):2482-93. Otodental syndrome. Bloch-Zupan A, Goodman JR. Orphanet J Rare Dis. 2006 Mar 21;1:5. Review. Abnormalities of tooth structure/Dentin • Dentinogenesis imperfecta with or without progressive hearing loss, DSPP • Osteogenesis imperfecta, with dentinogenesis imperfecta COL1A1, COL1A2, CRTAP, LEPRE1, SERPINF1, SERPINH1, SP7, OSX … – Type I (B) and (C) 10% – Type III 50% – Type IV 80% OI Dentin dysplasia type I (short roots), AD Dentin dysplasia type II, AD Kantaputra P, Tanpaiboon P, Porntaveetus T,Ohazama A, Sharpe P, Rauch A, Hussadaloy A, Thiel CT. 2011. The smallest teeth in the world are caused by mutations in the PCNT gene. Am J Med Genet Part A 9999A: 1–6. Goldblatt syndrome: #184260 SPONDYLOMETAPHYSEAL DYSPLASIA WITH DENTINOGENESIS IMPERFECTA – Short stature, narrow thorax, pectus carinatum, Joint hyperextensibility, mesomelia 48 Gorlin Collection. Ehlers Danlos syndrome. Type VII (dermatosparaxis). Patient at 12 years. Oral cavity showing prolapse of lips and discolored permanent teeth. ADAMTS2, procollagen protease, AR , 5q35.3 49 Gorlin Collection. Ehlers Danlos syndrome. Type VII (dermatosparaxis). Patient at 12 years. Oral cavity showing prolapse of lips and discolored permanent teeth. ADAMTS2, procollagen protease, AR , 5q35.3 Amelogenesis imperfecta Hypoplasia Hypomineralised Hypomature From: J Dent Res. 2010 October; 89(10): 1024–1038. doi: 10.1177/0022034510375829 Copyright/License ► << Prev Figure 8. Next >> Amelogenesis and proteins Request permission to reuse Figure 8. AMELX ENAM MMP20 KLK4 WDR72 FAM83H DLX3 FAM20A TP63 CNNM4 ROGDI C4orf26 SLC24A4 Xp22.3-p22.1 4q21 11q22.3-q23 19q13.3-q13.4 15q21.3 8q24.3 17q21.3-q22 17q24.2 3q27-q29 2q11.2 16p13.3 4q21.1 14q32.12 LAMB3 1q32 Major activities of maturation stage ameloblasts. (A) Calcium (Ca2+) and phosphate (H2PO4- and HPO42-) ions are transported and add to the width and thickness of existing calcium hydroxyapatite crystals generating hydrogen ions (H+). (B) Enamel proteins are cleaved by kallikrein (KLK4) and reabsorbed into the cells, possibly with the assistance of WDR72. (C) Magnesium ions (Mg2+) are potentially removed from the matrix by CNNM4. (D) Carbonic anhydrase II (CA2) catalyzes the combination of carbon dioxide (CO2) and water (H2O) to form a bicarbonate ion (HCO3-) and a hydrogen ion. The H+ is removed from the cell, possibly by the action of a sodium (Na+) and hydrogen ion http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3086535/figure/fig8-0022034510375829/ Regulation of dental enamel shape and hardness. Simmer JP, Papagerakis P, Smith CE, Fisher DC, Rountrey AN, Zheng L, Hu JC. J Dent Res. 2010 Oct;89(10):1024-38. Page 1 sur 2 Amelogenin, X-linked Enamelin Matrix metalloprotéinase 20 Kallikreine 4 WD repeat domain 72 Family with sequence similarity 83 member H Distalless Homeobox 3 Family with sequence similarity 20 member A Transformation-related protein 63 Cyclin M4 Rogdi homolog (Drosophila) Chromosome 4 open reading frame 26 Potassium dependent sodium/calcium exchanger A subunit of Laminin-5 AMEL X AI hypoplasia - Thin enamel Rough surface - Pits - Stripes - Normal and abnormal enamel - Banding pattern - Normal Dentine - Deletions and Mutations in the signal peptide coding region J Dent Res. 2004 May;83(5):378-83. Amelogenin p.M1T and p.W4S mutations underlying hypoplastic X-linked amelogenesis imperfecta.Kim JW, Simmer JP, Hu YY, Lin BP, Boyd C, Wright JT, Yamada CJ, Rayes SK, Feigal RJ, Hu JC. Am J Med Genet A. 2009 Aug;149A(8):1698-705. A large X-chromosomal deletion is associated with microphthalmia with linear skin defects (MLS) and amelogenesis imperfecta (XAI).Hobson GM, Gibson CW, Aragon M, Yuan ZA, Davis-Williams A, Banser L, Kirkham J, Brook AH. • deletion encompassing the entire AMELX gene, female patient MMP20 Homozygous mutation c.389C>T 444C>T ; c.389C>T ; p.T130I Homozygous and compound heterozygous MMP20 mutations in amelogenesis imperfecta. Gasse B, Karayigit E, Mathieu E, Jung S, Garret A, Huckert M, Morkmued S, Schneider C, Vidal L, Hemmerlé J, Sire JY, Bloch-Zupan A. J Dent Res. 2013 Jul;92(7):598-603. Homozygous and compound heterozygous MMP20 mutations in amelogenesis imperfecta. Gasse B, Karayigit E, Mathieu E, Jung S, Garret A, Huckert M, Morkmued S, Schneider C, Vidal L, Hemmerlé J, Sire JY, Bloch-Zupan A. J Dent Res. 2013 Jul;92(7):598-603. A: Normal enamel in a control tooth showing characteristic HunterSchreger bands. B: AI enamel of the first upper molar from the hypoplastic area. The darker layer corresponds to prenatal enamel. C: Enlargement of the prenatal enamel from the darker area in B. D: Enlargement of the postnatal enamel from the darker area in B. E: Enamel crystal alignment within the enamel rods of the control tooth. F: Enamel crystal perpendicular direction within the rods in the AI tooth. G: Presence of inter-rod enamel in the normal tooth. H: Absence or loss of interprismatic enamel in the AI tooth. Scale bars: A,B: 500 µm; C-H: 20 µm. Homozygous and compound heterozygous MMP20 mutations in amelogenesis imperfecta. Gasse B, Karayigit E, Mathieu E, Jung S, Garret A, Huckert M, Morkmued S, Schneider C, Vidal L, Hemmerlé J, Sire JY, Bloch-Zupan A. J Dent Res. 2013 Jul;92(7):598-603. An autosomal recessive cone-rod dystrophy associated with amelogenesis imperfecta. Michaelides M, Bloch-Zupan A, Holder GE, Hunt DM, Moore AT. J Med Genet. 2004 Jun;41(6):468-73. Mutations in CNNM4 cause Jalili syndrome, consisting of autosomalrecessive cone-rod dystrophy and amelogenesis imperfecta. Parry DA, Mighell AJ, El-Sayed W, Shore RC, Jalili IK, Dollfus H, BlochZupan A, Carlos R, Carr IM, Downey LM, Blain KM, Mansfield DC, Shahrabi M, Heidari M, Aref P, Abbasi M, Michaelides M, Moore AT, Kirkham J, Inglehearn CF. Am J Hum Genet. 2009 Feb;84(2): 266-73. AI and nephrocalcinosis Dr Marie-Claude ADDOR, Service de Génétique Médicale, CH-1011 CHUV Lausanne • • • Nephrocalcinosis (enamel renal syndrome) caused by autosomal recessive FAM20A mutations. Jaureguiberry G, De la Dure-Molla M, Parry D, Quentric M, Himmerkus N, Koike T, Poulter J, Klootwijk E, Robinette SL, Howie AJ, Patel V, Figueres ML, Stanescu HC, Issler N, Nicholson JK, Bockenhauer D, Laing C, Walsh SB, McCredie DA, Povey S, Asselin A, Picard A, Coulomb A, Medlar AJ, Bailleul-Forestier I, Verloes A, Le Caignec C, Roussey G, Guiol J, Isidor B, Logan C, Shore R, Johnson C, Inglehearn C, AlBahlani S, Schmittbuhl M, Clauss F, Huckert M, Laugel V, Ginglinger E, Pajarola S, Spartà G, Bartholdi D, Rauch A, Addor MC, Yamaguti PM, Safatle HP, Acevedo AC, MartelliJúnior H, dos Santos Netos PE, Coletta RD, Gruessel S, Sandmann C, Ruehmann D, Langman CB, Scheinman SJ, Ozdemir-Ozenen D, Hart TC, Hart PS, Neugebauer U, Schlatter E, Houillier P, Gahl WA, Vikkula M, Bloch-Zupan A, Bleich M, Kitagawa H, Unwin RJ, Mighell A, Berdal A, Kleta R. Nephron Physiol. 2012;122(1-2):1-6. Epileptic encephalopathy and amelogenesis imperfecta (Kohlschütter-Tönz syndrome) ROGDI on chromosome 16p13.3 Schossig-Zschocke AJHG 2012 Tuberous sclerosis, TSC1 9q34, TSC2 16p13.3, AD Tooth eruption/resorption anomalies Pycnodysostosis CTSK cathepsin K, AR, 1q21 Short stature Deformity of the skull (including wide sutures), maxilla and phalanges (acroosteolysis), osteosclerosis, and fragility of bone Micrognathia Narrow palate Delayed eruption of deciduous teeth Persistence of deciduous teeth Delayed eruption of permanent teeth Hypodontia Caries, enamel hypoplasia Primary failure of tooth eruption, PTHR1, parathyroid hormone receptor-1 gene, AD, 3p22-p21.1 - Hypodontia - Posterior openbite - Ankylosis of deciduous teeth - No responsive to orthodontic treatment Keratinisation defect • Papillon Lefevre syndrome • #245000 – 11q14.1-q14.3 – Cathepsin C Homozygous for the R272P mutation likely to cause a problem in protein folding parents are heterozygous for the same mutation Hypophosphatasia ALPL Childhood form - Mutation A 159T (c.526G>A), heterozygous, from paternal origin, moderate forms - Mutation c.648+1G>A, heterozygous, from maternal origin, severe forms Orodental phenotype and genotype findings in all subtypes of hypophosphatasia. Reibel A, Manière MC, Clauss F, Droz D, Alembik Y, Mornet E, Bloch-Zupan A. Orphanet J Rare Dis. 2009 Hypophosphatasia ALPL, Infantile form Familial expansile osteolysis TNFRSF11A, RANK, AD 18q21.1-22 Genodermatosis/Cancer Gorlin syndrome • #109400 – 9q31, 9q22.3 - PTCH Quality of care • • • • • • • Multidisciplinary teams Baby to Adults Prevention Evidence based dentistry Guidelines Chair side management and care to general anaesthesia or conscious sedation Partnership with local treating dental practitioners http://mediad4.u-strasbg.fr/register.php IGBMC Virginie LAUGEL, Marie PASCHAKI, Julia MARRIE, Arnaud LANGER, Pascal DOLLE André HANAUER, Raymond RIPP, Guillaume BERTHOMMIER, Olivier POURQUIE La plate-forme technologie: Biopuces Christelle THIBAULT La plateforme de Bioinformatique Intégrative et Génomique Jean MULLER, Olivier POCH ICS C. PILGRAM, J.L. MANDEL, T SORG, P. CHAMBON, S MULLER, Y HERAULT ICH, UCL Patrizzia FERRETTI, Patimaporn PUNGCHANCHAIKUL Collaborations Hélène DOLLFUS, Corinne STOETZEL, Mégana PRASAD, Mathilde HUCKERT, Laboratoire de génétique médicale, INSERM-UMR 1112, Université de Strasbourg Faculté de Médecine Uwe STRAHLE, Cristelle ETARD Karlsruher Institut für Technologie (KIT), Institut für Toxikologie und Genetik (ITG), Germany Jean Yves SIRE, Research group "Evolution & Développement du Squelette-EDS", UMR 7138-SAE, Université Pierre et Marie Curie, Paris, France Ann HUYSSEUNE, Université de Ghent, Belgique HUS – API - PHRC 2008-2013 Amélogenèse imparfaite Centre de référence des manifestations odontologiques des maladies rares Marie Cécile MANIÈRE, François CLAUSS, Sébastien TROESTER, Marzena SWITALA, Elodie FEISTHAMMEL Service de Biophysique et Médecine Nucléaire P. CHOQUET, A. CONSTANTINESCO Service de génétique médicale Hélène DOLLFUS DRC Hélène KUISSU, Naoual YAHMI Egide PHC France KKU Thaïlande Patimaporn PUNGCHANCHAIKUL, Morkmued SUPAWICH IFRO Institut Français de Recherche en Odontologie INTERREG, Offensive Sciences A27, Orodental manifestations of rare diseases, Vanessa STOEHR, Ute Moog, Anna WOLFF, Unikinikum Heidelberg, Stéphanie FEIERABEND, Uniklinikum Freiburg Fondation maladies rares Région Alsace, Ministerium für Wissenschaft, Forschung und Kunst Baden Württemberg, Ministerium für Bildung, Wissenschaft, Weiterbildung und Kultur des Landes RheinlandPfalz, Université de Strasbourg, Hôpitaux Universitaires de Strasbourg, CERBM – IGBMC, EA3949 - Laboratoire de Génétique Médicale, Universitätsklinikum Freiburg, Universität Heidelberg, Hypophosphatasie Europe, O b e r r h e i n i s c h e Zahnärtztegesellschaft [email protected]