Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
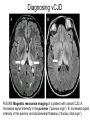
Prion Lecture Week 11 Medical Microbiology SBM 2044 Prion Diseases • Prion diseases are associated with the accumulation in the CNS of an abnormal form of a host protein called the prion protein, PrP • Infectious agent = an abnormally folded, degradation-resistant form of the PrP protein • The disease is rare, fatal and rapidly progressive neurodegenerative diseases that occur in humans and other animal species • They share common recognisable neuropathologic features: – presence of small vacuoles within the neuropil, produces a spongiform appearance, – neuronal loss, – glial cell proliferation. Human Prion Diseases • Kuru • Creutzfeldt-Jakob disease (CJD) • Variant Creutzfeldt-Jakob disease (variant CJD) • Gerstmann-Straussler-Scheinker syndrome (GSS) • Fatal familial insomnia (FFI) Prions • Proteinaceous infectious particle = small infectious agent that consists of protein but lacks nucleic acid • PrP is a host protein, and the sole constituents of prions • The gene encoding PrP is found in the genomes of all humans and animals – expressed in most human tissues, mainly in the CNS • Function of PrP ? ? – copper-binding protein, in cellular response to oxidative stress – long-term memory PrPSc • PrPSc is a pathological protease-resistant form of PrP – First isolated from the brains of animals with scrapie • Prion diseases: PrP PrPSc – a conformational change in PrP from a predominantly -helical form to -sheet CJD • 1 case per 1 million population worldwide each year • Sporadic disease • Age at onset usually 55-65 years old • Mean survival of 5 months • Britain – patients in their 20s… variant CJD? • Clinical manifestations: – – – – Rapidly progressive dementia and myoclonus, memory loss, judgment difficulties cognitive disturbances death within 1 year Diagnosing CJD Magnetic resonance imaging of a patient with CreutzfeldtJacob disease. A. Increased signal intensity in the putamen and head of the caudate (arrows). B. Bilateral parieto-occipital cortical hyperintensity (arrows). Diagnosing CJD Pathologic specimen from a patient with CJD demonstrating spongiform changes and neuronal loss. Treatment and Prevention • No specific treatment • Prions are resistant to routine sterilization and decontaminating procedures • PrPSc can be activated by – prolonged autoclaving (at 121°C and 15 psi for 4 ½ hours – immersion in 1 N NaOH (for 30 mins, repeat 3x) • Prohibition on ruminant-derived products for all animal feed Variant CJD • Bovine-to-human transmission of bovine spongiform encephalopathy (BSE), known as “mad cow disease”. • PrP proteins show different glycosylation pattern and electrophoretic mobility, than other prion diseases • Appear in the late 1990s, following epizootic of BSE in Britain – may be caused by changes in the rendering process of bovine byproducts, use for cattle feed (cannibalism) • Average age at onset 30 years old, a mean survival of 14 months Epidemiology of CJD Annual frequency of bovine spongiform encephalopathy (BSE) and variant CJD (vCJD) in Great Britain, 1988– 2003. Damaging Effects of vCJD • Clinical manifestations depend of the locations of PrPSc accumulation in the NS • All forms of CJD are uniformly fatal • vCJD patients have: – prominent sensory disturbances (on the face, limbs and torso) – psychiatric symptoms – depression – apathy, anxiety, intermittent delusions and psychosis – abnormalities of gaze Diagnosing vCJD • Laboratory and imaging studies are unhelpful • CSF shows no cells, mild elevation of CSF protein • MRI can be normal. But many patients present with signal hyperintensity in the pulvinar • Presence of plaques which stain for PrPSc throughout cerebrum and cerebellum, basal ganglia and thalamus • PrPSc has also been identified in tonsil biopsy tissue (nonneural tissue! ! !) Diagnosing vCJD FIGURE Magnetic resonance imaging of a patient with variant CJD. A. Increased signal intensity in the pulvinar (“pulvinar sign”). B. Increased signal intensity in the pulvinar and dorsomedial thalamus (“hockey stick sign”). Other Human Prion diseases • Gerstmann-Straussler-Scheinker syndrome (GSS) – autosomal dominant pattern of inheritance – progressive cerebellar degeneration accompanied by dementia • Fatal familial insomnia (FFI) – rapidly fatal midlife disease – a mean survival of 13 months – progressive insomnia, often with a dreamlike confusional state during waking hours – inattention, memory loss, confusion, hallucinations – myoclonus, ataxia and spasticity in later stage of disease – dysautonomia (hyperhidrosis, hyperthermia, tachycardia and hypertension) and endocrine disturbances ( adrenocorticotropic hormone secretion, cortisol secretion) Review of the Course Content • • • • • Microbes – Man interactions Week 1-3 Medical Bacteriology Week 4-6 Medical Virology Week 7-10 Medical Mycology Week 11-12 Emerging infectious diseases & biological agents of warfare Week 13 • Introduction to the medical diagnostics in microbiology Week 14 Quiz on all of the following topics: 1. Vaccines and Immunisations 2. Medical Diagnostics in Microbiology 3. Emerging and Reemerging Infectious Diseases 4. Biological Weapons 5. Prions • • Day/Date: Thursday 13th March 2008, Time: 2:30-3:30pm Happy studying!!